Introduction
Polycystic ovary syndrome (PCOS) is a prevalent
endocrine disorder affecting 10–13% of women worldwide (1). PCOS is clinically defined by
ovulatory dysfunction, hyperandrogenism, polycystic ovarian
morphology and chronic inflammation (2). Notably, PCOS is associated with
suboptimal pregnancy outcomes, primarily attributed to compromised
oocyte quality, oxidative stress and mitochondrial dysfunction,
with oocyte quality being a critical determinant of female
fertility. Granulosa cells (GCs), key somatic support cells for
oocytes, rely on functionally intact mitochondria to support their
growth, proliferation and steroidogenic capacity (3). Mitochondrial dysfunction in GCs has
emerged as a pivotal contributor to impaired oocyte maturation and
reduced developmental competence in patients with PCOS (4,5).
Studies have shown that in patients with PCOS, GCs exhibit
decreased mitochondrial membrane potential (MMP) and impaired
oxidative phosphorylation; these abnormalities lead to decreased
ATP synthesis and antioxidant enzyme activity, and oxidative stress
(6,7).
Research has shown that GCs from patients with PCOS
exhibit reduced mitochondrial biosynthesis, aberrant fission and
fusion dynamics, decreased MMP and activated mitochondrial
autophagy (8). Mitochondrial
fragmentation, resulting from imbalanced fission and fusion
processes, is considered a prerequisite for the initiation of
mitophagy (9,10). Notably, dynamin-related protein 1
(DRP1) serves a key role in mitochondrial dynamics; DRP1 mediates
mitochondrial fission by interacting with mitochondrial outer
membrane adaptor proteins, such as mitochondrial fission 1 protein
(FIS1) and mitochondrial fission factor (11). These interactions lead to the
formation of a division ring, which drives the fission process.
Dysregulation of mitochondrial dynamics and abnormalities in the
mitophagy pathway have been implicated in the pathogenesis of
various diseases, including cardiovascular disorders, cancer and
neurodegenerative conditions (12–14).
The sirtuin (SIRT) family comprises conserved class III histone
deacetylases. Among them, SIRT3 is primarily localized to the
mitochondria and has a pivotal role in preserving mitochondrial
function (15). Bugga et al
(16) revealed that SIRT3 may
alleviate cardiac mitochondrial dysfunction by activating
AMP-activated protein kinase (AMPK)/peroxisome
proliferator-activated receptor γ coactivator (PGC)-1α signalling.
SIRT3 knockdown impaired mitochondrial biogenesis and dynamics,
whereas its overexpression upregulated the expression of
mitochondrial DNA-encoded genes, increased superoxide dismutase 2
(SOD2) activity and altered mitochondrial dynamics. Additionally,
SIRT3 overexpression increased the expression of mitochondrial
biogenesis-related genes and proteins (PGC-1α and TFAM) (16). However, to the best of our
knowledge, the role of SIRT3 in regulating mitochondrial dynamics
in the GCs of patients with PCOS has not been reported. Therefore,
investigating regulators of mitochondrial dynamics may offer
insights for the development of novel therapeutic strategies for
treating PCOS.
Melatonin (MT), a pineal hormone and potent
antioxidant, regulates circadian rhythms and scavenges free
radicals (17). The level of MT is
greater in follicular fluid (FF) than in serum and it is markedly
increased during follicular maturation. Notably, low levels of MT
have been observed in the FF of patients with PCOS (18). Liu et al (19) reported that MT ameliorated cerebral
ischaemia/reperfusion injury through the activation of SIRT3
signalling. Similarly, Wu et al (20) demonstrated that MT inhibited
excessive mitochondrial autophagy in H9c2 cells through the MT
membrane receptor 2 (MT2)/SIRT3/FoxO3a axis, thereby alleviating
hypoxia/reoxygenation injury. However, the underlying molecular
mechanism of MT in PCOS remains to be elucidated.
On the basis of accumulating evidence that
mitochondrial dysfunction serves a pivotal role in PCOS
pathogenesis and that SIRT3 acts as a key regulator of
mitochondrial homeostasis, it was hypothesized that SIRT3-mediated
disruption of mitochondrial dynamics in GCs could contribute to the
development of PCOS, and that MT may ameliorate this dysfunction by
upregulating SIRT3 expression. Specifically, the present study
aimed to investigate the following: i) Whether GCs derived from
patients with PCOS exhibit SIRT3 downregulation and aberrant
mitochondrial dynamics; ii) whether SIRT3 overexpression can rescue
mitochondrial function in a PCOS-like cellular model; iii) whether
MT treatment can mimic the effects of SIRT3 overexpression; and iv)
whether the protective effects of MT are dependent on SIRT3.
Elucidation of these mechanisms may advance clinical diagnostics,
treatment paradigms and assisted reproductive outcomes in patients
with PCOS.
Materials and methods
Patients
Women who attended and were treated at the
Reproductive Medicine Centre of Hebei Reproductive Health Hospital
(Shijiazhuang, China) between June 2021 and June 2024 were included
in the present study. Patients with PCOS (n=36) were diagnosed
according to the Rotterdam criteria (21), and at least two of the following
features were detected: Oligo-ovulation/anovulation, biochemical or
clinical hyperandrogenism, and polycystic ovarian morphology on
ultrasound. The exclusion criteria for the control and PCOS groups
included the following: i) Ovarian cysts; ii) diminished ovarian
reserve; iii) endometriosis; iv) hydrosalpinx; v) chromosomal
abnormalities; or vi) chronic systemic conditions that could
influence gene expression in GCs. The control group (n=41) included
age-matched women with tubal factor infertility or who were
undergoing fertility treatment due to male factor infertility, and
with no endocrine abnormalities.
Isolation of cumulus GCs
Oocyte-cumulus complexes were retrieved by
transvaginal ultrasound-guided follicular aspiration 36 h after
human chorionic gonadotropin administration. The FF was collected
from the same follicular aspirates after removal of the
oocyte-cumulus complexes. Oocyte-cumulus complexes were
subsequently washed in several flat dishes with G-Mopse PLUS
(Vitrolife AB) to remove residual mural GCs, haemocytes and
cellular debris. Cumulus complexes were identified under a
microscope and mechanically flattened in Petri dishes. Cumulus GCs
(CGCs) surrounding the oocytes were then carefully dissected.
Transmission electron microscopy
(TEM)
Following centrifugation at 400 × g for 10 min at
room temperature, the CGCs were processed using standard
ultrastructural techniques. The samples were fixed in 2.5%
glutaraldehyde at 4°C for 2 h and pre-embedded in 1% agarose (Ted
Pella Inc.) to stabilize the pellet. Secondary fixation was
performed with 1% osmium tetroxide for 2 h at room temperature,
followed by sequential dehydration in a graded ethanol series. The
specimens were then infiltrated and embedded in
Araldite® epoxy resin. For TEM analysis, CGC samples
from three randomly selected patients per group were examined.
Ultrathin sections (60 nm) were prepared and allowed to air-dry
overnight at room temperature. For staining, the sections were
double-stained with 2% uranyl acetate in saturated alcoholic
solution and lead citrate, each for 15 min at room temperature.
Multiple ultrathin sections were prepared from each sample, and
imaging areas were randomly selected at low magnification by
electron microscopy technicians who were blinded to group
allocation. Representative images were subsequently captured at
higher magnifications to observe mitochondrial morphology and
autophagic structures. A total of 10–15 cells were randomly
selected from each sample. Autophagosomes were counted at the same
magnification, and the results are presented as the mean ± SD for
each group.
Cell culture
The human GC line KGN (cat. no. CL-0603), derived
from a GC tumour, was obtained from Procell Life Science &
Technology Co., Ltd. Both CGCs and KGN cells were maintained in
DMEM/F-12 (cat. no. D6421; Sigma-Aldrich; Merck KGaA) supplemented
with 10% heat-inactivated foetal bovine serum (FBS; cat. no.
A5256701; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin under standard culture conditions (37°C, 5%
CO2). Cellular morphology and proliferation kinetics
were monitored daily using phase contrast microscopy. At 80–90%
confluence, adherent cells were dissociated with trypsin-EDTA and
subcultured at a 1:3 ratio to maintain logarithmic growth.
Treatment of KGN cells
KGN cells were stimulated with dihydrotestosterone
(DHT; cat. no. 15874; Cayman Chemical Company) at various
concentrations (0, 10, 50, 100, 500, 1,000 and 2,000 nM) at 37°C
for 24 h, and the optimum concentration was selected for subsequent
experiments. In addition, KGN cells were exposed to MT (cat. no.
M5250; Sigma-Aldrich; Merck KGaA) at concentrations of 0, 0.1 and
10 pM, 1 and 100 nM at 37°C for 48 h, and the optimum concentration
(10 pM) was selected for subsequent experiments. In addition, KGN
cells were first exposed to 100 nM DHT for 24 h followed by
treatment with or without MT (10 pM) for an additional 48 h at
37°C. To investigate the effect of MT on SIRT3 expression in human
ovarian CGCs, CGCs obtained from control subjects and patients with
PCOS were cultured in vitro. The cells were treated with 10
pM MT at 37°C for 48 h, after which, the cells were harvested, and
SIRT3 protein expression levels were measured using western blot
analysis.
Cell transfection
Cells were transfected with SIRT3 overexpression
plasmids (pCMV6-Entry backbone, Myc-DDK-tagged) purchased from
OriGene Technologies, Inc. (cat. no. RC200190), or with an empty
pCMV6-Entry plasmid in the negative control (NC) group, using
Lipofectamine® 3000 (cat. no. L3000015; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, when the KGN cell density reached 70–80%,
transient transfection was performed; 4 µg plasmid solution was
diluted with serum-free medium and gently mixed, and P3000
(supplied with Lipofectamine 3000) was added. Lipofectamine 3000
was equilibrated to room temperature and gently mixed with
serum-free Opti-MEM® (cat. no. 31985; Gibco; Thermo
Fisher Scientific, Inc.), followed by incubation at room
temperature for 5 min to allow complex formation. Subsequently, the
plasmid DNA solution was diluted in an equal volume of Opti-MEM and
combined with the lipid mixture (Lipofectamine 3000 and Opti-MEM).
The DNA-lipid complexes were allowed to form through a 10-min
incubation at room temperature, after which, they were added
dropwise to the cell culture dish using a sterile pipette. The dish
was gently agitated to ensure an even distribution of the
transfection mixture and was then transferred to a humidified 37°C
incubator containing 5% CO2 for 4 h. Following this
incubation period, the transfection medium was replaced with
complete growth medium supplemented with 10% FBS, and the cells
were maintained under standard culture conditions for an additional
24 h prior to an efficiency assessment through western blot
analysis. For the inhibitor experiments, the cells were treated
with the SIRT3 inhibitor 3-TYP (50 µM cat. no. 29660; Cayman
Chemical Company) for 4 h at 37°C.
Chemical treatment
KGN cells were treated with 100 nM DHT at 37°C for
24 h to establish the DHT-induced model, followed by transfection
with the SIRT3 overexpression plasmid for at 37°C for 4 h. After
transfection, the medium was replaced, and the cells were cultured
for an additional 48 h. Then, chloroquine (CQ; 50 µM; cat. no.
C6628; Sigma-Aldrich; Merck KGaA) was added for 4 h at 37°C before
cell collection. The protein expression levels of LC3 and P62 were
subsequently detected by western blotting.
Cell counting Kit (CCK)-8 assay
A CCK-8 assay (cat. no. CA1211; Beijing Solarbio
Science & Technology Co., Ltd.) was used to measure the
viability of CGCs, according to the manufacturer's instructions.
Briefly, the cells were seeded on 96-well plates at a density of
2×104 cells/well, after which, 10 µl CCK8 reagent was
added to the culture medium and the plate was incubated for 1.5 h.
Finally, the absorbance was detected at 450 nm using a microplate
reader (SpectraMax® iD3; Molecular Devices, LLC). The
inhibition rate was calculated as follows: Inhibition rate (%)=[(OD
control-OD treatment)/(OD control-OD blank)] ×100. All CCK-8 assays
were performed in triplicate and repeated three times.
ELISA
The collected FF was centrifuged at 400 × g for 10
min at room temperature, after which the cell-free supernatant was
collected as the FF sample and stored at −80°C for subsequent
assays. The MT content of FF was measured using a Human MT ELISA
Kit (cat. no. EH3344; Wuhan Fine Biotech Co., Ltd.), according to
the manufacturer's instructions.
Measurement of reactive oxygen species
(ROS) levels
KGN cells (1×104 cells/well) were loaded
with 10 µmol/l dichlorodihydrofluorescein diacetate (DCFH-DA;
Beyotime Biotechnology) by incubation at 37°C for 20 min.
Subsequently, the fluorescence intensity was quantified using a
SpectraMax iD3 multifunctional microplate reader with
excitation/emission wavelengths set at 488/525 nm.
MMP detection
The MMP of KGN cells was measured using the JC-1
fluorescent probe (Cayman Chemical Company). KGN cells
(1×105 cells/well) were seeded in 24-well plates. After
24 h of culture, the cells were subjected to cell transfection or
exposure to 10 pM MT as aforementioned, and then labelled with 10
µmol/l JC-1 for 20 min in an atmosphere containing 5%
CO2 at 37°C. Fluorescence images were captured using an
Image Xpress confocal microscope (Molecular Devices, LLC) to
visualize both green (monomeric) and red (aggregated) fluorescence.
For each group, 5–6 fields were randomly selected at low
magnification by researchers who were blinded to the experimental
groups to avoid selection bias. The total red and green
fluorescence intensities in each field were measured using ImageJ
software (version 1.51; National Institutes of Health), and MMP is
expressed as a red/green fluorescence ratio. The data were obtained
from at least three independent experiments.
Mito-Tracker Red CMXRos staining
Active mitochondria were labelled using Mito-Tracker
Red CMXRos (Invitrogen; Thermo Fisher Scientific, Inc.). KGN cells
(1×105 cells/well) were seeded in 24-well plates. After
24 h of culture, the cells were subjected to cell transfection or
exposure to 10 pM MT as aforementioned, and were then incubated
with 10 µM MitoTracker Red CMXRos staining solution at 37°C for 20
min. After incubation, the cells were washed three times with PBS
(Wuhan Servicebio Technology Co., Ltd.) and maintained in fresh
culture medium. The fluorescence intensity was detected with an
Xpress confocal microscope. For each group, 5–6 fields were
randomly selected at low magnification by researchers who were
blinded to group allocation. The mean red fluorescence intensity
per field was measured using ImageJ software (version 1.51) to
determine the mitochondrial mass. The data were obtained from at
least three independent experiments.
Western blotting
CGCs and KGN cells were homogenized in RIPA buffer
(cat. no. RW0001; Hebei Ruipate Bio & Technology Co., Ltd.) and
the protein concentration was determined using a BCA kit. Protein
samples (5–8 µg) were then separated by SDS-PAGE on 10% gels and
transferred to a polyvinylidene fluoride membrane (Sigma-Adrich;
Merck KGaA). Subsequently, a blocking buffer containing 5% non-fat
dried milk (Inner Mongolia Yili Industrial Group Company Ltd.) was
used for 1.5 h at room temperature. The membrane was then incubated
with primary antibodies against SIRT3 (1:1,000; cat. no. ab217319;
Abcam), optic atrophy 1 (OPA1; 1:500; cat. no. ET1705-9; Huabio),
DRP1 (1:1,000; cat. no. IPB0168; Baijia), phosphorylated (P)-DRP1
(Ser616) (1:1,000; cat. no. AF8470; Affinity
Biosciences), mitofusin 2 (MFN2; 1:1,000; cat. no. 9482S; Cell
Signaling Technology, Inc.), FIS1 (1:500; cat. no. ET7109-17;
Huabio), P62 (1:2,000; cat. no. PM045; MBL International Co.), LC3
(1:500; cat. no. PM036; MBL International Co.), and β-actin
(1:5,000; cat. no. AC026; ABclonal Biotech Co., Ltd.) overnight at
4°C. Following the primary antibody incubation, the membrane was
incubated with HRP-goat anti-rabbit IgG (1:3,000; cat. no. RS0002;
ImmunoWay Biotechnology Company) or HRP-goat anti-mouse IgG
(1:3,000; cat. no. S1001; Hebei Ruipate Bio & Technology Co.,
Ltd.). The protein bands were visualized using an ECL detection
reagent (Shanghai Yeasen Biotechnology Co., Ltd.) on the Minichemi
320 chemiluminescence imaging system (Beijing Sage Creation Science
Co., Ltd.) and protein levels were semi-quantified with ImageJ
(version 1.51) software.
Statistical analysis
Data are presented as the mean ± SD for normally
distributed data, or as median (interquartile range) for
non-normally distributed data. Statistical analyses were performed
using SPSS 26.0 software (IBM Corp.). Normality of the data
distribution was assessed using the Shapiro-Wilk test. For data
following a normal distribution with homogeneous variances,
parametric tests were applied; comparisons between two groups were
performed using an independent sample t-test, whereas comparisons
among multiple groups were performed using one-way ANOVA followed
by Tukey's post hoc test. For data that did not follow a normal
distribution or with unequal variances, nonparametric tests were
applied; comparisons between two groups were performed using
Mann-Whitney U test, whereas comparisons among multiple groups were
performed using Kruskal-Wallis test followed by the Dunn-Bonferroni
test for post hoc pairwise comparisons. Each experiment was
repeated using at least three independent primary CGC samples
(biological replicates, n ≥3 per group), and each biological
replicate was measured in two technical replicates. Due to inherent
limitations in obtaining primary CGCS, sample sizes varied across
different detection markers; detailed sample sizes are provided in
the Results section and corresponding figure legends. P<0.05 was
considered to indicate a statistically significant difference.
Results
Comparison of clinical characteristics
and hormone levels between the PCOS and control groups
The clinical characteristics of the study population
are summarized in Table I. A total
of 36 women with PCOS and 42 age-matched controls were included in
the analysis. No significant differences were observed between the
two groups regarding age, body mass index, baseline oestradiol and
progesterone levels, metaphase II oocyte rate, fertilization rate
or high-quality embryo rate. However, distinct endocrine profiles
were observed: Compared with in the control group, the PCOS group
had significantly elevated baseline testosterone levels, increased
luteinizing hormone (LH) concentrations and a significantly higher
LH/follicle-stimulating hormone (FSH) ratio. Conversely, basal
serum FSH and prolactin levels were notably lower in patients with
PCOS.
 | Table I.Comparison of the general condition
between the two patient groups. |
Table I.
Comparison of the general condition
between the two patient groups.
| Characteristic | CON group
(n=41) | PCOS group
(n=36) | P-value |
|---|
| Age, years | 31.17±3.30 | 31.47±3.31 | 0.691 |
| BMI,
kg/m2 | 24.40±3.95 | 26.24±4.85 | 0.07 |
| Basal FSH,
IU/l | 7.07±1.67 | 6.22±1.81 | 0.034a |
| Basal LH, IU/l | 4.28±1.84 | 6.08±3.25 | 0.003a |
| LH/FSH | 0.63±0.29 | 1.00±0.55 | 0.001a |
| Basal E2,
pg/ml | 38.96±12.36 | 36.80±12.73 | 0.453 |
| Basal P, ng/ml | 0.62±0.37 | 0.64±0.88 | 0.871 |
| PRL, ng/ml | 15.21±7.83 | 11.27±4.89 | 0.011a |
| Basal T, ng/ml | 0.38±0.19 | 0.48±0.24 | 0.049a |
| MII rate | 0.84±0.10 | 0.77±0.22 | 0.076 |
| Fertilization
rate | 0.87±0.15 | 0.87±0.20 | 0.845 |
| Excellent embryo
rate | 0.60±0.27 | 0.57±0.24 | 0.591 |
CGCs from patients with PCOS exhibit
mitochondrial dysfunction and aberrant autophagy activation
To investigate mitochondrial dynamics in CGCs,
western blot analysis was performed on samples obtained from 9
patients with PCOS and 9 control patients. Owing to the limited
protein yield from CGCs, the effective sample size for certain
markers was smaller than that of the initial cohort (n<9), and
specific sample sizes are indicated in the corresponding results.
The protein levels of DRP1, MFN2, OPA1, FIS1 and P-DRP1
(Ser616) were detected by western blotting. The results
revealed that the expression levels of MFN2 and OPA1 were
significantly lower in the PCOS group than in the control group
(Fig. 1A). Conversely, the
expression level of FIS1 and the ratio of P-DRP1
(Ser616)/total DRP1 were significantly increased in the
PCOS group. These findings indicated a pronounced imbalance in
mitochondrial dynamics in CGCs derived from patients with PCOS,
characterized by increased fission and decreased fusion. In
addition, SIRT3 protein levels were significantly lower in the CGCs
from the PCOS group compared with those from the control group, as
determined by western blotting, suggesting that SIRT3
downregulation may contribute to mitochondrial dysfunction in PCOS
GCs (Fig. 1C).
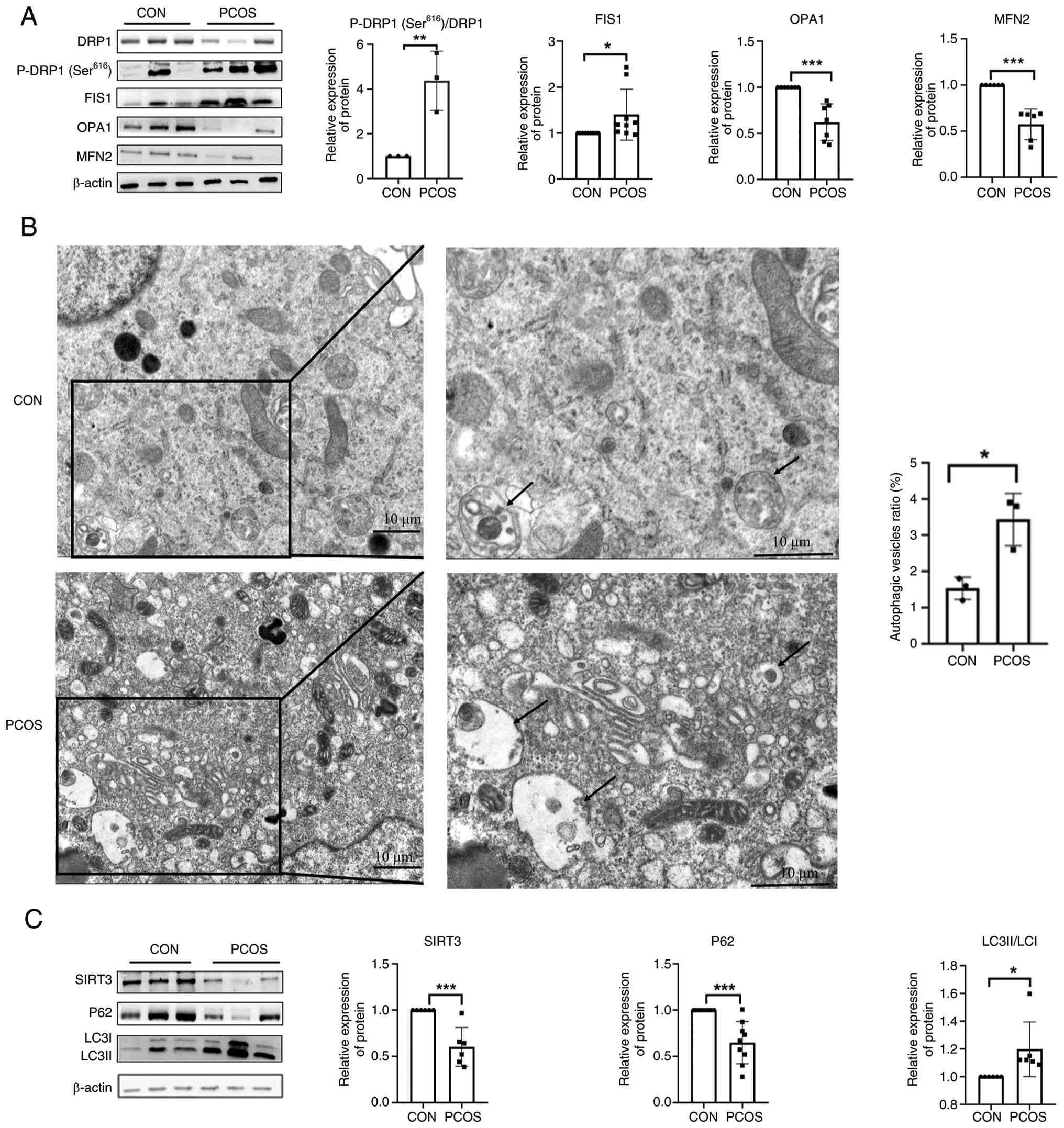 | Figure 1.CGCs from patients with PCOS exhibit
mitochondrial dysfunction and aberrant autophagy activation. (A)
Abnormal expression of mitochondrial fusion/fission proteins in the
CGCs of patients with PCOS. Protein levels of MFN2 (n=6 patients),
OPA1 (n=7 patients), FIS1 (n=9 patients), DRP1 (n=6 patients) and
P-DRP1 (Ser616) (n=3 patients) were detected by western
blotting in CGCs from patients with PCOS and controls. (B)
Ultrastructural evidence of autophagic activation and the
autophagic vesicle ratio in CGCs from patients with PCOS and
controls. Transmission electron micrographs showing increased
autophagosome density (black arrows) in PCOS CGCs (n=3) compared
with controls (n=3). Scale bar, 10 µm. (C) Western blot analysis of
autophagy-related markers in CGCs from patients with PCOS and
controls. Western blot analysis revealed reduced SIRT3 (n=6) and
P62 (n=9) expression, with a concomitant increase in the LC3-II/I
ratio (n=6) in PCOS CGCs compared with the controls. *P<0.05,
**P<0.01, ***P<0.001. CON, control; CGC, cumulus granulosa
cell; DRP1, dynamin-related protein 1; FIS1, mitochondrial fission
1 protein; MFN2, mitofusin 2; OPA1, optic atrophy 1; P-,
phosphorylated; PCOS, polycystic ovary syndrome; SIRT3, sirtuin
3. |
To evaluate autophagic activity in the CGCs of
patients with PCOS, autophagic vesicles were observed under
electron microscopy (Fig. 1B). The
results revealed an increased number of autophagic vesicles within
the ovarian CGCs of patients with PCOS, suggesting that autophagy
may be increased in the CGCs of patients with PCOS.
Furthermore, the expression levels of
autophagy-related markers, LC3 and P62, were analysed. Compared
with those in the control group, the PCOS group exhibited a
significantly increased LC3II/I ratio and significantly decreased
P62 expression levels (Fig. 1C),
indicating that autophagic activity was enhanced in the CGCs of
patients with PCOS.
SIRT3 overexpression enhances
mitochondrial function and suppresses autophagy in DHT-treated KGN
cells
The granulosa-like tumour cell line KGN, which
retains its steroidogenic capacity and serves as a robust model for
human GC research (22), was used
to establish an in vitro PCOS model through DHT induction, a
widely adopted approach in PCOS studies (23–25).
To optimize the treatment conditions, KGN cells were exposed to
increasing concentrations of DHT (0–2,000 nM) for 24 h. CCK-8
analyses revealed that DHT inhibited cell viability in a
concentration-dependent manner (Fig.
2A). Notably, treatment with 100 nM DHT reduced cell viability
by ~50%. Western blot analysis was then performed to assess the
protein expression levels of SIRT3, mitochondrial fission/fusion
markers (OPA1, DRP1 and MFN2) and P62 in KGN cells treated with
various concentrations of DHT (Fig.
2B). Consistent with the expression patterns observed in GCs
from patients with PCOS, 100 nM DHT markedly downregulated SIRT3,
DRP1, OPA1, MFN2 and P62 expression compared with in the untreated
control group. Therefore, this concentration was selected for
subsequent experiments.
![SIRT3 overexpression enhances
mitochondrial function and suppresses autophagy in KGN cells. (A)
Effect of different concentrations of DHT on KGN cell viability
(n=6). (B) Expression of mitochondrial dynamics-related proteins
SIRT3, DRP1, P-DRP1 (Ser616), MFN2 and OPA1 in response
to 0, 100, 250 and 500 nM DHT, as detected by western blotting
(n=6). (C) Western blot analysis to assess the expression levels of
SIRT3 in SIRT3-overexpressing KGN cells, confirming transfection
efficiency. (D) Mitochondrial membrane potential in KGN cells
overexpressing SIRT3 (n=5) Scale bars, 100 µm. (E) Effect of SIRT3
overexpression on the number of mitochondria (n=6). Scale bars, 100
µm. Expression levels of (F) mitochondrial dynamics-related
proteins [SIRT3, DRP1, P-DRP1 (Ser616), MFN2 and OPA1],
and (G) LC3 and P62 were detected by western blotting (n=6). (H)
Expression levels of LC3-II and P62 in the presence of CQ was
detected by western blotting (n=6). *P<0.05, **P<0.01 vs. CON
group; #P<0.05 vs. DHT group, $$P<0.01
vs. NC group; &&P<0.01 vs. CON + CQ group,
@P<0.05 vs. DHT + CQ group. CON, control; CQ,
chloroquine; DHT, dihydrotestosterone; DRP1, dynaminrelated protein
1; FIS1, mitochondrial fission 1 protein; MFN2, mitofusin 2; NC,
negative control; OPA1, optic atrophy 1; P-, phosphorylated; SIRT3,
sirtuin 3.](/article_images/mmr/34/2/mmr-34-02-13937-g01.jpg) | Figure 2.SIRT3 overexpression enhances
mitochondrial function and suppresses autophagy in KGN cells. (A)
Effect of different concentrations of DHT on KGN cell viability
(n=6). (B) Expression of mitochondrial dynamics-related proteins
SIRT3, DRP1, P-DRP1 (Ser616), MFN2 and OPA1 in response
to 0, 100, 250 and 500 nM DHT, as detected by western blotting
(n=6). (C) Western blot analysis to assess the expression levels of
SIRT3 in SIRT3-overexpressing KGN cells, confirming transfection
efficiency. (D) Mitochondrial membrane potential in KGN cells
overexpressing SIRT3 (n=5) Scale bars, 100 µm. (E) Effect of SIRT3
overexpression on the number of mitochondria (n=6). Scale bars, 100
µm. Expression levels of (F) mitochondrial dynamics-related
proteins [SIRT3, DRP1, P-DRP1 (Ser616), MFN2 and OPA1],
and (G) LC3 and P62 were detected by western blotting (n=6). (H)
Expression levels of LC3-II and P62 in the presence of CQ was
detected by western blotting (n=6). *P<0.05, **P<0.01 vs. CON
group; #P<0.05 vs. DHT group, $$P<0.01
vs. NC group; &&P<0.01 vs. CON + CQ group,
@P<0.05 vs. DHT + CQ group. CON, control; CQ,
chloroquine; DHT, dihydrotestosterone; DRP1, dynaminrelated protein
1; FIS1, mitochondrial fission 1 protein; MFN2, mitofusin 2; NC,
negative control; OPA1, optic atrophy 1; P-, phosphorylated; SIRT3,
sirtuin 3. |
To investigate the functional role of SIRT3 in
DHT-treated KGN cells, SIRT3 overexpression was used to assess
mitochondrial function. Firstly, successful SIRT3 overexpression in
KGN cells was confirmed, with protein levels increasing ~2-fold
relative to those in the NC cells (Fig. 2C), thereby validating the
transfection efficiency.
SIRT3 modulates MMP and dynamics (26); therefore, to evaluate the
regulatory role of SIRT3 in mitochondrial function, three
experimental groups were analysed: Untreated control, DHT-treated
and DHT + SIRT3 groups. The MMP was assessed using JC-1 staining.
Cells with a low MMP contained JC-1 monomers and exhibited green
fluorescence signals, whereas those with a high MMP showed red
fluorescence signals due to JC-1 polymer formation. JC-1 staining
revealed a significant reduction in the MMP in DHT-treated cells
compared with that in the control group, whereas this effect was
partially reversed by SIRT3 overexpression (Fig. 2D). By contrast, MitoTracker
staining revealed no significant differences in mitochondrial
fluorescence intensity across groups, suggesting that mitochondrial
mass remained unchanged (Fig.
2E).
DHT treatment significantly reduced the protein
levels of the mitochondrial fusion proteins OPA1 and MFN2 and
increased the P-DRP1 (Ser616)/DRP1 ratio (Fig. 2F). Notably, SIRT3 overexpression in
DHT-treated cells upregulated OPA1 and MFN2 expression (P<0.05
vs. DHT) and decreased the P-DRP1 (Ser616)/DRP1 ratio,
suggesting that SIRT3 may enhance mitochondrial function. The
expression levels of autophagy-associated proteins were assessed by
western blotting. DHT treatment significantly reduced the protein
levels of the autophagic substrate P62 (Fig. 2G), indicating increased autophagic
flux. Notably, SIRT3 overexpression in DHT-treated cells
significantly increased P62 expression compared with DHT alone,
suggesting impaired autophagic flux. LC3-II levels showed an
increasing trend in both DHT and DHT + SIRT3 groups, but the
changes were not statistically significant. As shown in Fig. 2G, overexpression of SIRT3 in
DHT-treated cells led to elevated P62 levels, which appears to
exert an opposite effect to DHT. It remains unclear whether SIRT3
overexpression inhibits autophagic flux, selectively regulates
mitophagy without affecting general autophagy, or induces
compensatory changes in P62 synthesis. To definitively address this
issue, autophagic flux was analysed in KGN cells using CQ. CQ
blocks lysosomal degradation, so the extent of LC3-II and P62
accumulation in the presence of CQ is proportional to autophagic
flux; a greater accumulation reflects higher autophagic flux. The
cells were divided into the following six groups: Control, control
+ CQ, DHT, DHT + CQ, DHT + SIRT3 and DHT + SIRT3 + CQ, and the
accumulation of LC3II and P62 in the presence of CQ was compared.
The accumulation of LC3II and P62 in the DHT + CQ group was
significantly greater than that in the control + CQ group,
confirming that DHT increased autophagic flux (Fig. 2H). Furthermore, the accumulation of
LC3II and P62 in the DHT + SIRT3 + CQ group was significantly lower
than that in the DHT + CQ group, indicating that SIRT3
overexpression inhibited the DHT-mediated increase in autophagic
flux.
MT upregulates SIRT3 in CGCs and
protects mitochondrial function in DHT-induced KGN cells
To assess MT levels in patients with PCOS, the
concentrations of MT in the FF were measured using ELISA. Compared
with in the FF from control patients, the FF from patients with
PCOS had significantly lower MT levels (172.7±51.63 vs.
205.02±59.87 pg/ml; Fig. 3A).
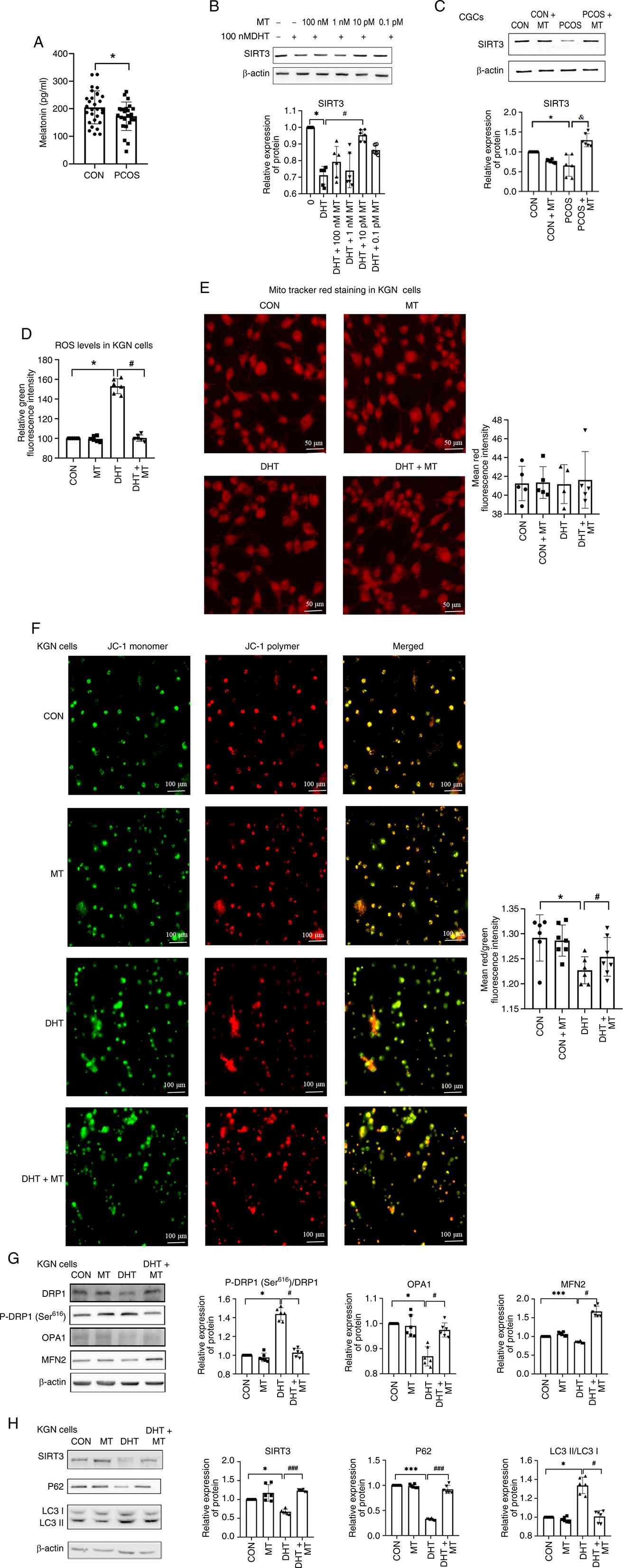 | Figure 3.MT upregulates SIRT3 in CGCs and
protects mitochondrial function in DHT-induced KGN cells. (A)
Comparison of MT levels in the follicular fluid between the two
groups of patients (n=29 vs. n=24). (B) KGN cells were cultured
with different concentrations of MT, and the expression of SIRT3
was compared (n=6). (C) Protein expression levels of SIRT3 in CGCs
of the four groups (n=6). (D) ROS levels in KGN cells from the four
groups. Dichlorodihydrofluorescein diacetate staining was performed
to measure the concentrations of ROS in those groups, and the
staining was detected using a microplate reader (n=6). (E) Changes
in fluorescence intensity were observed using MitoTracker Red dye
(n=5). Scale bar, 50 µm. (F) MMP detected with Image Xpress
confocal microscopy and analysis of MMP by JC-1 staining in CGCs
from the CON group, CON + MT group, DHT group and DHT + MT group
(n=6). Scale bar, 100 µm. Western blot analysis of (G) DRP1,
P-DRP1Ser616, OPA1 and MFN2, and (H) SIRT3, P62 and LC3
expression in the CON group, CON + MT group, DHT group and DHT + MT
group (n=6). *P<0.05, ***P<0.001 vs. CON group;
#P<0.05, ###P<0.001 vs. DHT group,
&P<0.05 vs. PCOS + MT. CON, control; CGC, cumulus
granulosa cell; DHT, dihydrotestosterone; DRP1, dynaminrelated
protein 1; FIS1, mitochondrial fission 1 protein; MFN2, mitofusin
2; MMP, mitochondrial membrane potential; MT, melatonin; OPA1,
optic atrophy 1; P-, phosphorylated; PCOS, polycystic ovary
syndrome; ROS, reactive oxygen species; SIRT3, sirtuin 3. |
To investigate the regulatory effect of MT on SIRT3
expression, KGN cells were cotreated with 100 nM DHT and various
concentrations of MT; the results revealed that treatment with 10
pM MT significantly increased SIRT3 expression compared with DHT
(Fig. 3B).
In human CGCs, SIRT3 protein levels were
significantly lower in PCOS-derived CGCs than in control CGCs. By
contrast, MT treatment of CGCs form patients with PCOS (PCOS + MT
group) restored SIRT3 expression to near-control levels (Fig. 3C).
To assess the effect of MT on mitochondrial
homeostasis, the intracellular ROS levels in DHT-induced KGN cells
were measured by DCFH-DA staining. Notably, ROS levels were
increased in the DHT group, whereas this increase was normalized by
cotreatment with MT (Fig. 3D).
Mito Tracker Red staining revealed no significant differences in
mitochondrial mass among the groups (control, control + MT, DHT and
DHT + MT; Fig. 3E). However, JC-1
staining revealed a decrease in the MMP in DHT-treated cells, which
was effectively restored by MT (Fig.
3F).
Western blot analysis of mitochondrial
dynamics-related proteins revealed that DHT increased P-DRP1
(Ser616) levels, and reduced MFN2 and OPA1 levels, all
of which were normalized by MT cotreatment (Fig. 3G). These results suggested that MT
may ameliorate the DHT-induced abnormalities in mitochondrial
dynamics.
DHT exposure decreased P62 and SIRT3 levels, and
increased the LC3-II/I ratio, whereas MT treatment reversed these
effects (Fig. 3H). Together, these
findings suggested that MT alleviates DHT-induced mitochondrial
dysfunction by modulating SIRT3 expression and regulating
mitophagy.
SIRT3 inhibition abrogates the effects
of MT on mito-chondrial dynamics and autophagy
To further verify whether MT regulates mitochondrial
dynamics by upregulating SIRT3 expression, KGN cells were treated
with or without DHT for 24 h, then treated with the SIRT3-specific
inhibitor 3-TYP for 4 h, followed by treatment with MT for 48 h.
KGN cells were divided into four groups: Control, DHT, DHT + MT and
DHT + MT + 3-TYP, and the expression levels of mitochondrial
dynamics-associated proteins were assessed by western blotting. The
results showed that the protein expression levels of OPA1 and MFN2
in the DHT + MT group were significantly higher than those in the
DHT group, but were significantly reduced after the addition of the
SIRT3 inhibitor 3-TPY (Fig. 4A).
The ratio of P-DRP1 (Ser616)/DRP1 in the DHT + MT group
was significantly lower than that in the DHT group; however, the
addition of the SIRT3 inhibitor did not alter this ratio. This
revealed that MT failed to reverse the DHT-induced abnormalities in
mitochondrial fusion when SIRT3 expression was inhibited,
suggesting that SIRT3 is essential for MT-mediated regulation of
mitochondrial fusion.
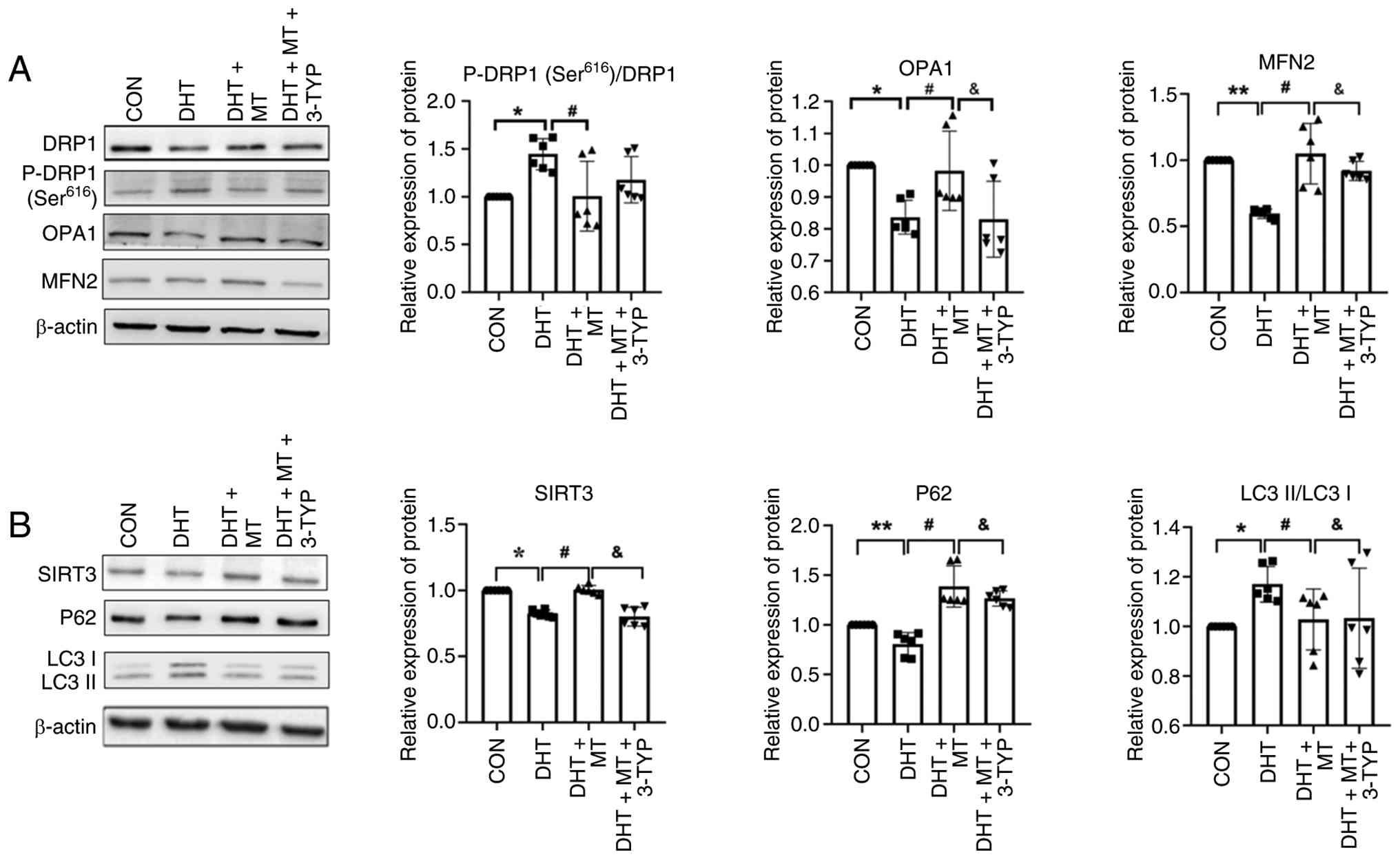 | Figure 4.A SIRT3 inhibitor abrogates the
effects of MT on mitochondrial dynamics and autophagy. Western blot
analysis of (A) DRP1, P-DRP1 (Ser616), OPA1 and MFN2
expression, and (B) SIRT3, P62 and LC3 expression (n=6). Data are
presented as the mean ± SD. *P<0.05, **P<0. vs. CON group;
#P<0.05 vs. DHT group; &P<0.05 vs.
DHT+MT group. CON, control; DHT, dihydrotestosterone; DRP1,
dynamin-related protein 1; MFN2, mitofusin 2; MT, melatonin; OPA1,
optic atrophy 1; P-, phosphorylated; SIRT3, sirtuin 3. |
To verify whether MT alleviates DHT-induced
autophagic dysfunction through upregulation of SIRT3 expression,
the expression of autophagy-related markers was assessed by western
blotting. Following SIRT3 inhibition, MT was unable to suppress
excessive autophagy (Fig. 4B).
These findings further support that MT may restore mitochondrial
dynamics and autophagic activity by increasing SIRT3
expression.
Discussion
The primary clinical manifestation in patients with
PCOS is anovulation, which primarily results from impaired
follicular development and arrested maturation. As critical
regulators of primordial follicle activation during folliculogenes
(27), ovarian GCs substantially
influence follicular initiation and maturation through the
functional integrity of their mitochondria. Previous investigations
have demonstrated marked mitochondrial dysfunction in CGCs derived
from patients with PCOS, characterized by a decreased MMP,
excessive accumulation of ROS and dysregulation of mitochondrial
biogenesis pathways (28,29). Given the essential role of CGC
mitochondria in coordinating follicular development and oocyte
competence, the precise pathophysiological mechanisms linking
PCOS-associated mitochondrial abnormalities to impaired oocyte
quality remain incompletely understood. This highlights the
critical need to elucidate the molecular cascades through which
PCOS compromises oocyte developmental potential while also
exploring innovative strategies to optimize reproductive outcomes
in this prevalent endocrine disorder.
Mitochondrial dynamics and damage exhibit a tightly
coupled bidirectional relationship (30). Mitochondrial impairment can disrupt
dynamic equilibrium, thereby compromising cellular energy
homeostasis, apoptotic regulation and other critical biological
processes, whereas aberrant mitochondrial dynamics may conversely
exacerbate mitochondrial damage (31). Upon phosphorylation at Ser616, DRP1
is activated and translocates to the mitochondria, where it binds
protein partners located on the outer mitochondrial membrane and
subsequently promotes mitochondrial fission (32). The findings of the present study
revealed significant downregulation of the mitochondrial fusion
regulators MFN2 and OPA1, indicating a reduction in mitochondrial
fusion in the CGCs of patients with PCOS. Activation of P-DRP1
(Ser616) facilitates the translocation of DRP1 to the mitochondria,
and an increased P-DRP1 (Ser616)/DRP1 ratio promotes
mitochondrial fission, leading to enhanced mitochondrial division,
thereby contributing to an imbalance in mitochondrial dynamics in
the context of PCOS. Notably, SIRT3 expression was markedly reduced
in the CGCs of patients with PCOS. Experimental evidence has
demonstrated that SIRT3 deficiency exacerbates mitochondrial
dysfunction, whereas its upregulation restores mitochondrial
dynamics in renal tubular systems (33). The pathological triad of oxidative
stress, mitochondrial injury and SIRT3 depletion has been
mechanistically linked, with SIRT3 supplementation showing
therapeutic potential in ameliorating renal damage in murine models
of acute kidney injury (34).
These findings collectively suggest that SIRT3 downregulation may
constitute a central mechanistic node underlying mitochondrial
dynamic disturbances in PCOS CGCs; thus, a novel therapeutic axis
for addressing ovarian dysfunction in this syndrome is
proposed.
Accumulating evidence highlights the critical role
of SIRT family proteins, particularly the understudied protein
SIRT3, in the pathogenesis of PCOS. While SIRT1 remains the focus
of current research, the current clinical data revealed significant
SIRT3 downregulation in patients with PCOS, which is consistent
with the findings in a previous study that SIRT3 knockdown in KGN
GCs mimics PCOS-associated glucose dysregulation and insulin
resistance (29). Utilizing a
DHT-induced in vitro model of PCOS, mitochondrial
dysfunction was observed, as characterized by reduced MMP and
elevated ROS levels, despite the unchanged mitochondrial mass. DHT
concurrently disrupted mitochondrial dynamics by increasing fission
and decreasing fusion while promoting mitophagy, as evidenced by
reduced P62 levels. These results reveal a dual mechanism whereby
hyperandrogenism induces mitochondrial pathology via SIRT3
deficiency-mediated fission and fusion imbalance and autophagic
flux arrest, positioning SIRT3 as a therapeutic target for
PCOS-related ovarian metabolic dysfunction.
The present study demonstrated that SIRT3
overexpression rescued mitochondrial homeostasis in KGN cells by
counteracting DHT-induced dysfunction. In KGN cells, SIRT3
overexpression significantly increased the MMP, while normalizing
mitochondrial dynamics by decreasing P-DRP1 (Ser616)
levels and increasing the expression of fusion regulators (MFN2 and
OPA1). Previous studies have shown that SIRT3 can influence
mitochondrial homeostasis by regulating upstream signalling
molecules (such as AMPK/PGC-1α) or antioxidant enzymes (such as
SOD2), thereby indirectly affecting the expression and function of
dynamic proteins (35,36). Therefore, the changes in total
protein levels observed in the current study may have resulted from
the overall regulatory network of SIRT3. These findings suggest
that SIRT3 mitigates PCOS-associated mitochondrial damage by
restoring fission and fusion equilibrium. SIRT3 overexpression
induced an increase in P62 levels, which indicates that it exerted
an opposite effect to DHT treatment alone, because DHT alone
decreased P62. To investigate whether the changes in P62 levels
reflect alterations in autophagic flux, cells were treated with CQ
and autophagic analysis was performed. The results of CQ treatment
indicated that SIRT3 overexpression significantly reduced LC3-II
and P62 accumulation in the DHT + SIRT3 + CQ group compared with in
the DHT + CQ group. Thus, changes in P62 levels did reflect
alterations in autophagic flux, and the findings indicated that
SIRT3 suppressed the DHT-enhanced autophagic flux. SIRT3 may impair
autophagic degradation efficiency, counteract DHT-induced
autophagic overactivation and reduce autophagic flux.
MT, a lipophilic hormone synthesized by the pineal
gland, exerts pleiotropic effects on cellular redox balance and
mitochondrial homeostasis through its membrane permeability
(37). Emerging evidence has
suggested that MT deficiency is involved in ovarian dysfunction,
with the present study confirming significantly reduced levels of
MT in the FF of patients with PCOS compared with those in the
control individuals. Mechanistically, exogenous MT treatment
increased SIRT3 expression in the CGCs of patients with PCOS. By
upregulating SIRT3 expression, MT reversed DHT-induced
mitochondrial damage in CGCs, as evidenced by the restored membrane
potential, attenuated oxidative stress, and normalized fission and
fusion dynamics. Crucially, MT suppressed excessive autophagy,
whereas the addition of a SIRT3 inhibitor abolished the beneficial
effects of MT on mitochondrial dynamics and mitophagy,
unequivocally indicating that SIRT3 is a key mediator of the
therapeutic effect of MT. These findings align with clinical
observations that MT supplementation alleviates PCOS-associated
metabolic disturbances (38),
positioning the MT-SIRT3 axis as a druggable pathway to counteract
androgen-driven mitochondrial pathology in patients with PCOS. The
present study revealed that MT significantly upregulated SIRT3
expression in CGCs; however, the upstream regulatory mechanism
remains to be elucidated. The biological effects of MT are
typically mediated through two pathways: One involves the
activation of downstream signalling cascades (such as PI3K/Akt and
PKA/CREB) via the membrane receptors MT membrane receptor 1 and
MT2, thereby regulating gene transcription (39,40);
the other involves its role as a potent antioxidant, which reduces
oxidative stress-induced damage to the SIRT3 protein by scavenging
free radicals, thus indirectly maintaining its expression levels
(41). Previous studies have
reported that MT can activate the SIRT3 signalling axis through MT2
to ameliorate cardiomyocyte hypoxia/reoxygenation injury (20), and can also exert antioxidative,
antiapoptotic and anti-inflammatory renoprotective effects via
SIRT3 activation (42,43). Future studies employing specific
receptor antagonists (such as luzindole) or antioxidant
interventions will help to elucidate the relative contributions of
these two mechanisms in GCs from patients with PCOS.
The present study identified the MT-SIRT3 axis as a
pivotal regulatory node in PCOS-associated mitochondrial
dysfunction, offering novel mechanistic insights into
hyperandrogenism-driven ovarian metabolic dysregulation. The
findings revealed that the SIRT3-dependent restoration of
mitochondrial dynamics and mitophagy is central to the therapeutic
effects of MT. However, the present study has several limitations.
First, while the use of cell lines is valuable for obtaining
mechanistic insights, it does not fully recapitulate the complex
in vivo environment. Owing to the limited protein yield from
primary CGCs, the sample size was relatively small, which may
affect the generalizability of the findings. The results should be
validated in future studies with larger sample sizes of primary
GCs. Second, the lack of validation in an in vivo animal
model of PCOS represents another key limitation. Although the
DHT-treated KGN cell line serves as a well-established in
vitro model for mechanistic studies, it cannot replicate the
complex systemic interactions that characterize the pathophysiology
of PCOS. Therefore, well-established PCOS animal models (such as
letrozole-induced or DHT-induced rodent models) should be employed
in future studies to validate the therapeutic efficacy of MT, and
assess its long-term effects on ovarian function, metabolic
parameters and reproductive outcomes. Third, MT is a circadian
hormone with a typical nocturnal secretion pattern and its levels
in FF may exhibit diurnal fluctuations. The current in vitro
experiments employed constant MT treatment, which does not mimic
the pulsatile or cyclical nature of MT exposure in vivo.
Future studies should investigate whether the timing of MT
administration affects its protective effects on the functions of
CGCs. Finally, the mechanistic interaction between MT and SIRT3 was
investigated primarily using pharmacological inhibition and
overexpression strategies, without direct molecular binding assays
(such as binding affinity determinations). Receptor blockade and
antioxidant interventions should be performed in future studies for
in-depth analysis. Moreover, molecular docking assays should be
prioritized to map MT-SIRT3 binding interfaces coupled with
multiomics profiling to decode downstream deacetylation targets.
These insights pioneer a paradigm shift towards precision
interventions targeting mitochondrial epigenetics in PCOS while
underscoring the importance of pharmacokinetic optimization to
harness the full therapeutic potential of this pathway.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Government Clinical
Medical Talent Training Program (grant nos. ZF2026372 and
ZF2026373).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CX, ZL, TC, HL, XZ, WY, JZ, DS and XW contributed to
study conception and design. CX and TC performed the experiments.
XZ, WY and JZ were responsible for the collection of CGCs. ZL and
HL performed the data collection. DS and ZL analysed the data. CX
and ZL wrote the first draft of the manuscript, and all authors
commented on previous versions of the manuscript. DS and XW confirm
the authenticity of all the raw data. All the authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Hebei Reproductive Health Hospital (approval no.
KYY-2021-LW-008; Shijiazhuang, China). Written informed consent was
obtained from each patient after a full explanation of the purpose
and nature of all the procedures used. The procedures of the
present study were in accordance with the ethical standards stated
in The Declaration of Helsinki (1964).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhang Y, Chen ZJ and Zhao H: Polycystic
ovary syndrome: A metabolic disorder with therapeutic
opportunities. Cell Metab. 37:1932–1949. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gao Y, Zou Y, Wu G and Zheng L: Oxidative
stress and mitochondrial dysfunction of granulosa cells in
polycystic ovarian syndrome. Front Med (Lausanne). 10:11937492023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alberico HC and Woods DC: Role of
granulosa cells in the aging ovarian landscape: A focus on
mitochondrial and metabolic function. Front Physiol. 12:8007392021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmalbrock LJ, Weiss G, Rijntjes E,
Reinschissler N, Sun Q, Schenk M and Schomburg L: Pronounced trace
element variation in follicular fluids of subfertile women
undergoing assisted reproduction. Nutrients. 13:41342021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sreerangaraja Urs DB, Wu WH, Komrskova K,
Postlerova P, Lin YF, Tzeng CR and Kao SH: Mitochondrial function
in modulating human granulosa cell steroidogenesis and female
fertility. Int J Mol Sci. 21:35922020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dabravolski SA, Nikiforov NG, Eid AH,
Nedosugova LV, Starodubova AV, Popkova TV, Bezsonov EE and Orekhov
AN: Mitochondrial dysfunction and chronic inflammation in
polycystic ovary syndrome. Int J Mol Sci. 22:39232021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Delcour C, Amazitl Patino LC, Magnin F,
Fagart J, Delemer B, Young J, Laissue P, Binart N and Beau I: ATG7
and ATG9A loss-of-function variants trigger autophagy impairment
and ovarian failure. Genet Med. 21:930–938. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang F, Han J, Wang X, Liu Y and Zhang Z:
Roles of HIF-1α/BNIP3 mediated mitophagy in mitochondrial
dysfunction of letrozole-induced PCOS rats. J Mol Histol.
53:833–842. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu H, Zang C, Yuan F, Ju C, Shang M, Ning
J, Yang Y, Ma J, Li G, Bao X and Zhang D: The role of FUNDC1 in
mitophagy, mitochondrial dynamics and human diseases. Biochem
Pharmacol. 197:1148912022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Poole LP and Macleod KF: Mitophagy in
tumorigenesis and metastasis. Cell Mol Life Sci. 78:3817–3851.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu S, Huang Z, Yin Z, Chen L, Ye L, Zhang
D and Bai X: Drp1 in Parkinson's disease: Mechanisms of disease
pathology and the promise of targeted therapeutic strategies. Life
Sci. 384:1240862026. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cai C, Guo Z, Chang X, Li Z, Wu F, He J,
Cao T, Wang K, Shi N, Zhou H, et al: Empagliflozin attenuates
cardiac microvascular ischemia/reperfusion through activating the
AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 52:1022882022.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu D, Liu R, Zhou Y, Zhang Z, Jiang X, Xu
J, Su A and Hu Z: FOXO3a-dependent up-regulation of HSP90
alleviates cisplatin-induced apoptosis by activating
FUNDC1-mediated mitophagy in hypoxic osteosarcoma cells. Cell
Signal. 101:1105002023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo X, Zhang Z, Gu J, Ke P, Liu J, Meng Y,
Zheng W, Que W, Fan R, Luo J, et al: FUDNC1-dependent mitophagy
ameliorate motor neuron death in an amyotrophic lateral sclerosis
mouse model. Neurobiol Dis. 197:1065342024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ansari A, Rahman MS, Saha SK, Saikot FK,
Deep A and Kim KH: Function of the SIRT 3 mitochondrial deacetylase
in cellular physiology, cancer, and neurodegenerative disease.
Aging Cell. 16:4–16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bugga P, Alam MJ, Kumar R, Pal S,
Chattopadyay N and Banerjee SK: Sirt3 ameliorates mitochondrial
dysfunction andoxidative stress through regulating mitochondrial
biogenesis and dynamics in cardiomyoblast. Cell Signal.
94:1103092022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sánchez A, Calpena AC and Clares B:
Evaluating the oxidative stress in inflammation: Role of melatonin.
Int J Mol Sci. 16:16981–17004. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H, Liu M and Zhang C: Women with
polycystic ovary syndrome (PCOS) have reduced melatonin
concentrations in their follicles and have mild sleep disturbances.
BMC Womens Health. 22:792022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu L, Chen H, Jin J, Tang Z, Yin P, Zhong
D and Li G: Melatonin ameliorates cerebral ischemia/reperfusion
injury through SIRT3 activation. Life Sci. 239:1170362019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu J, Yang Y, Gao Y, Wang Z and Ma J:
Melatonin attenuates anoxia/reoxygenation injury by inhibiting
excessive mitophagy through the MT2/SIRT3/FoxO3a signaling pathway
in H9c2 cells. Drug Des Devel Ther. 14:2047–2060. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rotterdam ESHRE/ASRM-Sponsored PCOS
Consensus Workshop Group, : Revised 2003 consensus on diagnostic
criteria and long-term health risks related to polycystic ovary
syndrome. Fertil Steril. 81:19–25. 2004. View Article : Google Scholar
|
|
22
|
Ji R, Jia FY, Chen X, Wang ZH, Jin WY and
Yang J: Salidroside alleviates oxidative stress and apoptosis via
AMPK/Nrf2 pathway in DHT-induced human granulosa cell line KGN.
Arch Biochem Biophys. 715:1090942022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang J, Huang H, Xiao M, Jiang X, Yang Y,
Huang M, Wang S, Zhu B and Zhao M: Erchen decoction ameliorates the
rat model of polycystic ovary syndrome by regulating the steroid
biosynthesis pathway. Phytomedicine. 143:1568522025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yi S, Zheng B, Zhu Y, Cai Y, Sun H and
Zhou J: Melatonin ameliorates excessive PINK1/Parkin-mediated
mitophagy by enhancing SIRT1 expression in granulosa cells of PCOS.
Am J Physiol Endocrinol Metab. 319:E91–E101. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Z, Wang RH and Wang KH: Formononetin
ameliorates polycystic ovary syndrome through suppressing NLRP3
inflammasome. Mol Med. 31:272025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu M, Xuan A, Zheng L, Li D, Chen C, Liu
H, Lu G, Cheng Z, Zou Y, Zhi S, et al: Novel coumarin derivative
SZC-6 as an allosteric activator of SIRT3 alleviates diabetic
kidney disease via the SIRT3-Foxo3a signaling axis. Free Radic Biol
Med. 240:29–45. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fiorentino G, Cimadomo D, Innocenti F,
Soscia D, Vaiarelli A, Ubaldi FM, Gennarelli G, Garagna S, Rienzi L
and Zuccotti M: Biomechanical forces and signals operating in the
ovary during folliculogenesis and their dysregulation: Implications
for fertility. Hum Reprod Update. 29:1–23. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie C, Lu H, Zhang X, An Z, Chen T, Yu W,
Wang S, Shang D and Wang X: Mitochondrial abnormality in ovarian
granulosa cells of patients with polycystic ovary syndrome. Mol Med
Rep. 29:272024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Q, Ren J, Wang F, Pan M, Cui L, Li M
and Qu F: Mitochondrial and glucose metabolic dysfunctions in
granulosa cells induce impaired oocytes of polycystic ovary
syndrome through Sirtuin 3. Free Radic Biol Med. 187:1–16. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Srinivasan S, Guha M, Kashina A and
Avadhani NG: Mitochondrial dysfunction and mitochondrial
dynamics-The cancer connection. Biochim Biophys Acta Bioenerg.
1858:602–614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Che L, Wu JS, Du ZB, He YQ, Yang L, Lin
JX, Lei Z, Chen XX, Guo DB, Li WG, et al: Targeting mitochondrial
COX-2 enhances chemosensitivity via Drp1-dependent remodeling of
mitochondrial dynamics in hepatocellular carcinoma. Cancers
(Basel). 14:8212022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mao R, He S, Lan J and Zhu WZ: Honokiol
ameliorates cisplatin-induced acute kidney injury via inhibition of
mitochondrial fission. Br J Pharmacol. 179:3886–3904. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Morigi M, Perico L, Rota C, Longaretti L,
Conti S, Rottoli D, Novelli R, Remuzzi G and Benigni A: Sirtuin
3-dependent mitochondrial dynamic improvements protect against
acute kidney injury. J Clin Invest. 125:715–726. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Y, Ge Y, Hua S, Shen C, Cai B and
Zhao H: Aloe-Emodin improves mitophagy in Alzheimer's disease via
activating the AMPK/PGC-1α/SIRT3 signaling pathway. CNS Neurosci
Ther. 31:e703462025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han X, Peng M, Wang X, Hu Y, Li G, Song Y,
Lv Y and Chen N: Role of the AMPK/PGC-1α/SIRT3-Mediated
mitochondrial dysfunction in the neurotoxicity of methanol. Mol
Neurobiol. 63:1342025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Megha KB, Arathi A, Shikha S, Alka R,
Ramya P and Mohanan PV: Significance of melatonin in the regulation
of circadian rhythms and disease management. Mol Neurobiol.
61:5541–5571. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Alizadeh M, Karandish M, Asghari
Jafarabadi M, Heidari L, Nikbakht R, Babaahmadi Rezaei H and
Mousavi R: Metabolic and hormonal effects of melatonin and/or
magnesium supplementation in women with polycystic ovary syndrome:
A randomized, double-blind, placebo-controlled trial. Nutr Metab
(Lond). 18:572021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Meng K, He Y, Wang H, Zhang Y and
Quan F: Melatonin stimulates STAR expression and progesterone
production via activation of the PI3K/AKT pathway in bovine theca
cells. Int J Biol Sci. 15:404–415. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shao X, Yang Y, Liu Y, Wang Y, Zhao Y, Yu
X, Liu J, Li YX and Wang YL.: Orchestrated feedback regulation
between melatonin and sex hormones involving GPER1-PKA-CREB
signaling in the placenta. J Pineal Res. 75:e129132023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reiter RJ, Sharma RN, Manucha W,
Rosales-Corral S, Almieda Chuffa LG, Loh D, Luchetti F, Balduini W
and Govitrapong P: Dysfunctional mitochondria in age-related
neurodegeneration: Utility of melatonin as an antioxidant
treatment. Ageing Res Rev. 101:1024802024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang C, Suo M, Liu L, Qi Y, Zhang C, Xie
L, Zheng X, Ma C, Li J, Yang J and Bu P: Melatonin alleviates
Contrast-Induced acute kidney injury by activation of Sirt3. Oxid
Med Cell Longev. 2021:66688872021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu S, Li L, Wu J, Fang H, Han Y, Huang Q,
Chen Z and Zeng Z: Melatonin attenuates Sepsis-induced
small-intestine injury by upregulating SIRT3-mediated
oxidative-stress inhibition, mitochondrial protection, and
autophagy induction. Front Immunol. 12:6256272021. View Article : Google Scholar : PubMed/NCBI
|














