Introduction
Studies on meningioma have been increasingly focused
on understanding their biology at the cellular level, particularly
through cell culture studies and animal models (1–6).
These models have allowed for investigations into the
characteristics, growth patterns and molecular mechanisms driving
meningioma development (1,3,5–12).
Cell culture models are typically used to explore
the genetic mutations and epigenetic modifications that contribute
to meningioma pathogenesis and formation (4–6).
Establishing primary meningioma cell cultures involves isolating
tumor cells from surgical specimens and maintaining them in
vitro under specific conditions that promote cell proliferation
and survival. Typically, meningioma cell cultures are prepared
using enzymatic digestion with various agents, such as collagenase
or trypsin, to dissociate the tumor tissue, followed by cultivation
in specialized media, including DMEM or RPMI, which are
supplemented with FBS, antibiotics and growth factors (1,2,4,6,13).
These cultures can be used to investigate cellular proliferation,
genetic mutations and tumor responses to therapeutic agents.
However, primary meningioma cultures frequently face challenges,
such as limited lifespan, heterogeneity, selection of potent cells
and difficulty in maintaining the original phenotype of the primary
tumor over time. To overcome these limitations, immortalized
meningioma cell lines have been developed through genetic
modification techniques, providing consistent and long-term models
for research (4–6,14,15).
Additionally, three-dimensional culture systems and spheroid models
are increasingly being utilized to optimally mimic the tumor
microenvironment (16).
Meningioma cell culture is a key method in tumor
research, allowing for the analysis of meningioma tumor cells ex
vivo. To the best of our knowledge, the physiology of cells,
which is influenced by conditions in the operating theatre,
transportation and storage from tissue removal to processing, is an
important but poorly documented factor in the literature,
especially regarding cells in primary cell culture.
Consequently, the present study used meningioma
samples post-surgery to investigate how storage time and further
cryopreservation treatment can affect the proliferative pattern of
primary cell cultures. To assess cytogenetics in these cell
cultures, fluorescence in situ hybridization (FISH) was
performed on all cultures, the results of which were compared with
those from native tumor tissue smears, as well as loss of
heterozygosity (LOH) analysis, to determine whether cell cultures
accurately represented the original tumor.
Materials and methods
Patient population and histology
From June 2022 to December 2022, 10 primary human
meningiomas were collected after surgery at the Department of
Neurosurgery at Saarland University (Homburg, Germany). Patients
undergoing elective surgery for suspected or previously diagnosed
meningioma were eligible for inclusion. Written informed consent
was obtained from each patient as per the protocol approved by the
Ethical Committee of the Medical Association of Saarland (approval
no. 02/20; Saarbrücken, Germany). A total of 10 tumor samples from
9 patients (3 male patients; 6 female patients) were included in
the present study. The mean age of patients at the time of surgery
was 75.2±9.4 years (range, 62–86 years). All patients had a primary
tumor and patients with recurrence were not included in the present
study. All tumors were classified according to the actual 2021
World Health Organization (WHO) classification of tumors of the
nervous system (17) by a
neuropathologist.
Cell culture and preparation
From each meningioma, fragments of the tumor were
placed by one individual in DMEM (Gibco; Thermo Fisher Scientific,
Inc.) with 1% penicillin/streptomycin directly after preparation in
the surgical field by a neurosurgeon in the operating theatre at
room temperature (22–28°C) before being transferred to the
neurosurgical laboratory with standard conditions as previously
described (1). A segment of the
tissue sample was prepared for culture on the same day as the
surgery took place, whilst another was stored overnight in DMEM
with 1% penicillin/streptomycin in a refrigerator (4–6°C). The
remainder of the tissue was cryopreserved in cryotubes in a freezer
at −80°C. For cell culture preparation, the tumor sample was minced
using a scalpel and small scissors. Cells were suspended in DMEM
containing 10% fetal calf serum (Thermo Fisher Scientific, Inc.),
1% non-essential amino acids and 1% penicillin/streptomycin, before
subsequently being distributed into two different 25-cm2
cell culture flasks for the next steps. The primary cultures were
incubated at 37°C with 5% CO2 in air and the medium was
changed twice per week.
Primary cell cultures at passage 0 (P0) were
established on the day of surgery and 1 day postoperatively, and
these were classified as culture groups A and B, respectively.
These cells from group A and B were stored overnight in DMEM with
1% penicillin/streptomycin in a refrigerator (4–6°C). Cultures from
group A and B were split into two passage 1 (P1) cultures. The
termination of the cell culture occurred when the cell culture
occupied the flask space completely or at least 10 days had passed.
One P1 culture was used for FISH, while the other culture was
frozen in liquid nitrogen after a cell count. The latter was then
thawed after 6–7 months and cultivated again at passage 2 (P2).
Cell counting, freezing and
thawing
The second culture flask from the first passage was
prepared for freezing in liquid nitrogen as aforementioned. The
medium in the appropriate culture flasks was first transferred to a
sterile round tube. The cell culture in each flask was then
incubated with 2 ml 0.05% trypsin-ethylenediaminetetraacetic acid
(EDTA) at 37°C in the incubator for ≥2 mins. Proteolysis was halted
by adding 2 ml of DMEM. The cell suspension was subsequently
transferred to a centrifuge tube and centrifuged at 37.02 × g for 5
min at room temperature with a brake. The supernatant was removed
and discarded, and the cell pellet was resuspended in 1 ml of fresh
medium.
For cell counting, 10 µl 0.4% trypan blue solution
was pipetted into a 1.5-ml tube. Cell suspension (10 µl) was added
and both liquids were mixed well. From this mixture, 10 µl was
placed onto a cell counting slide. The LUNA-II™ cell counter
(Aligned Genetics, Inc.) was used to analyze the sample, and
provided the total cell count, along with the number of live and
dead cells, and the viability ratio.
A special freezing medium was used for freezing of
the cells. This was prepared from 10 ml dimethyl sulfoxide (DMSO)
and 90 ml DMEM, aliquoted into 5-ml portions in round tubes and
stored at −20°C. DMSO protects cells from mechanical damage due to
ice crystals during freezing. The added medium was centrifuged at
37.02 × g for 5 min at room temperature with a brake. The
supernatant was pipetted out and discarded. The pellets were then
resuspended in 1 ml freezing medium and transferred to cryotubes,
which were placed inside a freezing box. The freezing container
filled with isopropanol allowed for controlled cooling of the cells
in the cryotubes, achieving a cooling rate of approximately −1°C
per min. The container was frozen overnight at −80°C. The following
day, the cryotubes were removed and transferred to liquid nitrogen
at −196°C, where the cells were stored for subsequent analysis.
After 6–7 months, the corresponding cryotubes were thawed and the
cells were recultivated at P2. The cultures from P2 were then
treated in the same manner as the primary cultures (as described
for P1) except for the stage involving the splitting of the initial
cultures into two separate cultures. Medium changes and termination
of the cultures occurred as soon as a dense cell lawn was observed
under the light microscope (Olympus CKX; Olympus Corporation) in
daily observations.
Preparation of dabbed slides
In addition to cell culture, one tumor fragment from
each sample at P1 and P2 was used for the preparation of dabbed
slides. To achieve this, a fragment of the tumor was dabbed onto
object slides coated with silane for 5 sec at room temperature, and
was fixed with DeLauney fixative, comprising 1:1 acetone/ethanol
with 0.05% trichloroacetic acid, at room temperature for 5 min.
Samples were stored at −20°C.
FISH
Primary tumor cells were dispersed with 0.05%
trypsin-EDTA and subsequently suspended by centrifugation at 30.85
× g for 8 min at room temperature. The supernatant was discarded,
and the pellet was treated with 0.075 M KCl solution at room
temperature for 5 min and resuspended. The cell suspension was
centrifuged once more at 30.85 × g for 8 min at room temperature.
The supernatant was once again removed and the cells were fixed
with methanol/acetic acid (3:1) for 1 h at room temperature.
Subsequently, the cell suspension was dropped onto the object dry
slides. Additionally, dabbed slides were used to compare both
methods as partially described previously (1).
The slides were treated with RNase A (Fisher
Scientific; Thermo Fisher Scientific, Inc.) for 30 min at 37°C and
placed three times in 2X saline sodium citrate (SSC) for 5 min at
room temperature. Cells were digested in 100 ml 0.01 M hydrogen
chloride with 10 mg pepsin (SERVA Electrophoresis GmbH) at 0.7 mA
for 1 min and 45 sec at 37°C. Slides were dipped in 1X PBS for 5
min at room temperature, 4% paraformaldehyde/1X PBS for 10 min at
room temperature for fixation and 1X PBS for 5 min at room
temperature. Samples were subsequently dehydrated in 70, 80 and 95%
ethanol and air-dried. Dual-probe hybridization was carried out
using locus-specific probes for band 36 on the short arm of
chromosome 1 (1p36; D6021-100-OG; MetaSystems) and band 11 on the
long arm of chromosome 22 (22q11; D5117-100-OG; MetaSystems).
The probes were then pipetted onto the slides and
denatured for 2 min at 75°C. Afterwards, samples were incubated
overnight at 37°C in a humidified chamber. Stringency washes were
performed in 0.4X SSC for 2 min at 72°C and 2X SSC/0.05% Tween-20
for 30 sec at room temperature. Following this, slides were
counterstained with DAPI antifade (Vectashield; Vector
Laboratories, Inc.; Maravai LifeSciences).
In total, ≥200 non-overlapping nuclei per sample
were evaluated according to the Hopman criteria (18) using an Olympus BX43 fluorescence
microscope (Olympus Corporation). Cut-offs for alterations were
determined by comparison with human lymphocytes as control samples
at 10% for deletions of 1p36 and 22q11.
LOH analysis
PCR-based microsatellite analysis was performed on
meningioma probes directly from the operating theatre (group A) in
a manner similar to the protocols previously described for
oligodendrogliomas, using different probes (18). For the PCR reaction 1.3 µl DNA, 1.0
µl primer mix (both forward and revers primer at a concentration of
20 µM; Eurofins Genomics), 10.2 µl water and 12.5 µl HotStarTaq
Master Mix Kit (Qiagen GmbH) were mixed (25.0 µl total reaction
volume). The PCR reaction was performed according to the protocol
developed by Hartmann et al (19).
For the investigation of the short arm of chromosome
1 (1p), the probes D1S 1608, D1S 1161 and D1S 1184 (created by
Qiagen GmbH for the Institute of Neuropathology, Saarland
University, Homburg, Germany), and a probe for the gene locus of
AT-rich interaction domain 1A were used. For the long arm of
chromosome 22 (22q), the following probes were used: D22S 445, D22S
684, D22S 268 and D22S 258 (created by Qiagen GmbH for the
Institute of Neuropathology, Saarland University). PCR products
were visualized on high-resolution Spreadex® EL 800 Wide
Mini gels (AL-Labortechnik & Diagnostik GmbH) using an Origins
electrophoresis system (AL-Labortechnik & Diagnostik GmbH) and
SYBR™-Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific, Inc.).
Gel electrophoresis was conducted for 90 min at 120 V in
TRIS-acetate-EDTA-buffer (cat. no. 42548.01; TAE buffer, SERVA
Electrophoresis GmbH) at 54°C. Subsequently, gels were washed once
with distilled water, stained with staining solution for 30 min at
room temperature [final buffer concentration, 0.8X TAE (cat. no.
42548.01; SERVA Electrophoresis GmbH), 0.8X Destaining Solution
(cat. no. 3037.01; SERVA Electrophoresis GmbH), 2X SYBR™ Gold] and
washed again with distilled water. The results were documented
using the ‘EOS Utility’ program (Canon, Inc.). Blood samples for
LOH analysis were obtained during clinical routine as a standard
procedure for neuropathological diagnostics and were stored at room
temperature in EDTA tubes for further analysis within 12–48 h.
Statistical analysis
The graphing and analysis of data were performed
using SPSS (version 27.0; IBM Corp.) and Microsoft Excel (Microsoft
365; Microsoft Corporation). The variables were initially tested
for normal distribution using the Shapiro-Wilk test. Normality was
assumed if the P-value exceeded the significance level. Fisher's
exact test was also used for calulations. Since some variables were
not normally distributed, the P-values were calculated using the
Mann-Whitney U test. Data are presented as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
General results
Primary cultures and native tissue samples from 10
meningiomas were established, derived from 3 male and 6 female
patients. Notably, 1 patient, designated patient 7, had two tumor
probes from different localizations (T9175 and T9177). These two
different tumors were surgically treated using two separate
surgical approaches and were treated as two separate tumor
pathologies in the clinical and scientific setting. All tumors
showed soft tumor tissue and could be cut for further preparation
of tumor samples. The mean age of patients at the time of surgery
was 75.2±9.4 years. Histopathological analysis revealed 8
meningiomas classified as WHO grade 1 and 2 meningiomas classified
as WHO grade 2. The tumors were located at the sphenoid wing (n=3),
convexity (n=2), parasagittal (n=3), posterior fossa (n=1) and
anterior skull base (n=1). Further details are provided in Table I.
 | Table I.Overview of the parameters of the
tumor samples. |
Table I.
Overview of the parameters of the
tumor samples.
| T-no. | Patient no. | Sex | Age, years | WHO grade | Cytogenetic
findings | FISH analysis | Localization |
|---|
| T9077 | 1 | Female | 82 | 1 | Diploid set of
chromosomes | Diploid set of
chromosomes | Convexity, on the
right, parasagittal |
| T9092 | 2 | Female | 65 | 1 | Diploid set of
chromosomes | Diploid set of
chromosomes | Sphenoid wing, on
the right |
| T9108 | 3 | Female | 74 | 1 | 22q- | 22q- | Convexity, on the
left, parietal |
| T9145 | 4 | Female | 84 | 1 | 1p-, 22q- | 1p-, 22q- | Parasagittal, on
the left, parietal |
| T9152 | 5 | Female | 86 | 1 | Diploid set of
chromosomes | Diploid set of
chromosomes | Sphenoid wing,
mediolateral |
| T9168 | 6 | Male | 75 | 1 | 1p-, 22q- | 1p-, 22q- | Sphenoid wing, on
the left |
| T9175 | 7 | Male | 62 | 1 | 22q- | 22q- | Posterior cranial
fossa, infratentorial |
| T9177 | 7 | Male | 62 | 2 | Diploid set of
chromosomes | 22q- | Cerebello-pontine
angle, on the left |
| T9181 | 8 | Male | 65 | 1 | 1p-, 22q- | 1p-, 22q- | Falx
cerebri/parasagittal, bifrontal |
| T9194 | 9 | Female | 84 | 2 | 1p- | 1p- | Frontobasal, on the
left |
Proliferation rate of P0 cultures in
cell culture
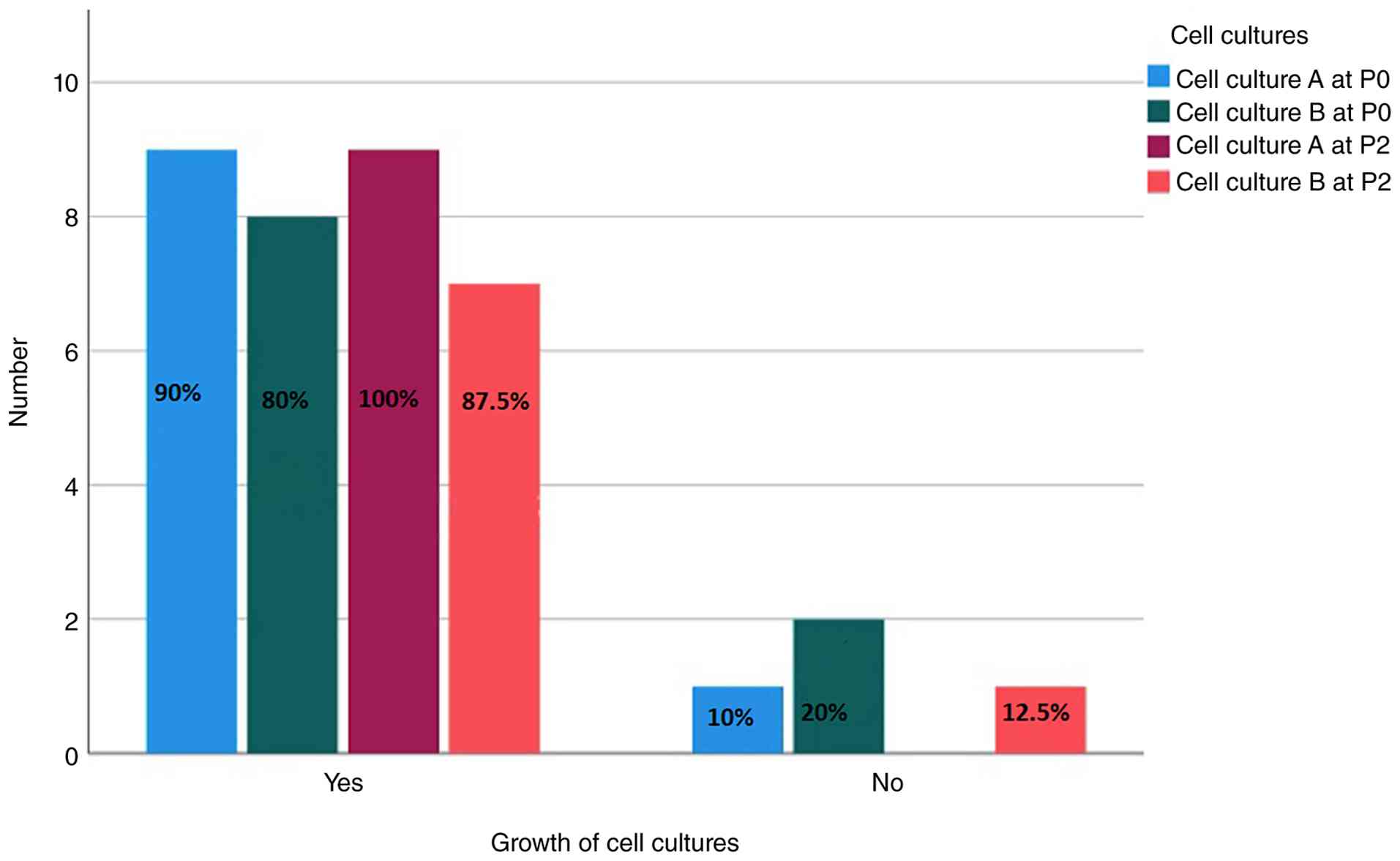
A total of 8 out of 10 meningioma samples showed
signs of proliferation in both cultures tested from the day of
surgery and from 1 day after surgery. In one case, only the sample
in group A P0 showed proliferation, whereas in another case, no
proliferation was observed in the group A P0 cell culture flask and
in the group B P0 cell culture flask. Overall, 17 out of 20 primary
cultures proliferated, whilst 3 did not show proliferation after 10
days of observation (Table II).
The proliferation rate was calculated as 85% for P0.
| Table II.Average duration (days) of cell
culture in groups A and B at passage 0 to splitting, passage 1, and
passage 2. |
Table II.
Average duration (days) of cell
culture in groups A and B at passage 0 to splitting, passage 1, and
passage 2.
|
| Passage 0 | Passage 1 | Passage 2 |
|---|
|
|
|
|
|
|---|
| Cell culture | Mean ± SD | N | Mean ± SD | N | Mean ± SD | N |
|---|
| A | 7.78±3.27 | 9 | 11.56±2.78 | 9 | 7.67±1.65 | 9 |
| B | 8.75±2.91 | 8 | 12.38±3.66 | 8 | 8.25±1.75 | 8 |
| Total | 8.24±3.05 | 17 | 11.94±3.15 | 17 | 7.94±1.67 | 17 |
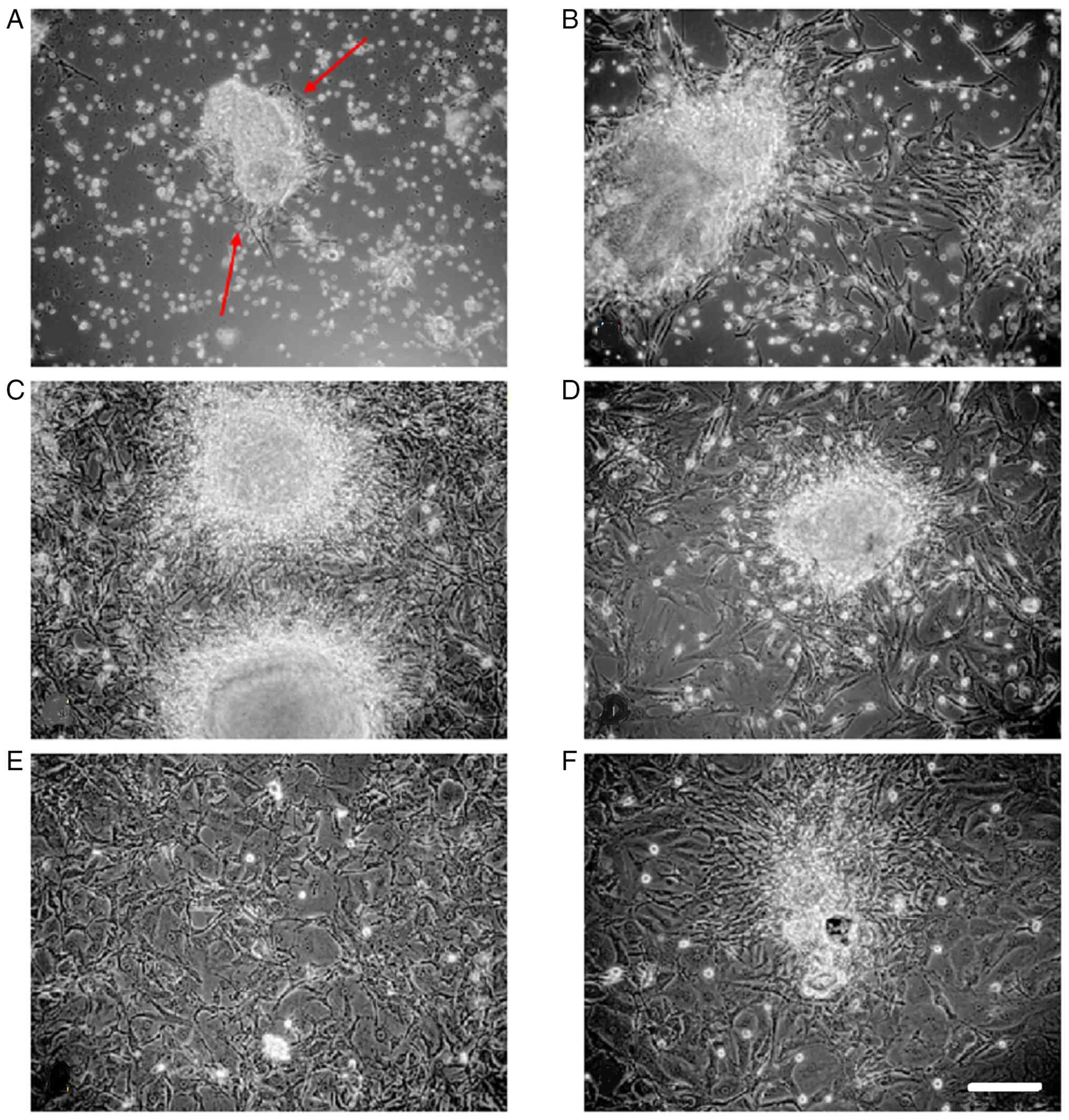
Culture on the day of surgery
There was sufficient cell proliferation in approach
A to transfer the P0 cultures into two P1 culture flasks, with 9
successful cultures among a total of 10 cultures (90%) in both. The
splitting process led to faster cell proliferation, as seen in
daily microscopic observations of the cell cultures, ensuring that
the P1 flasks contained enough cells to use one culture for FISH
and freeze the other in liquid nitrogen. Representative images of
cell culture are shown in Fig.
1.
Culture on the first postoperative
day
For approach B, widespread proliferation of
meningioma cells was observed in 8 out of 10 cultures (80%) at P0,
allowing for further processing. Cultures initiated on the first
postoperative day exhibited lower proliferation rates compared with
those established on the day of surgery. However, this difference
was not statistically significant (P>0.35) (Fig. 2; Table II).
Average time to cell outgrowth in
primary cultures
Cell outgrowth from tissue was observed after an
average of 1.4±0.9 days. Cultures from group A showed initial cell
proliferation after an average of 1.3±1.0 days, whereas cultures in
group B exhibited proliferation after an average of 1.5±0.9 days
(data not shown). The difference between these two groups was not
statistically significant.
Splitting of P0 cultures
Splitting of cultures at P0 could be performed after
8.24±3.05 days on average. Culture group A was split after an
average of 7.78±3.27 days, whilst culture group B was split after
8.75±2.91 days (Table II). This
indicated that group B cultures were split nearly a day later on
average; however, this difference was not statistically
significant.
Duration until culture termination for
FISH
The average duration before termination of primary
(P0 and P1) cultures for FISH was 11.94±3.15 days. Cultures from
group A were terminated after an average of 11.56±2.78 days,
whereas group B cultures were terminated after 12.38±3.66 days. The
difference was not statistically significant (Table II).
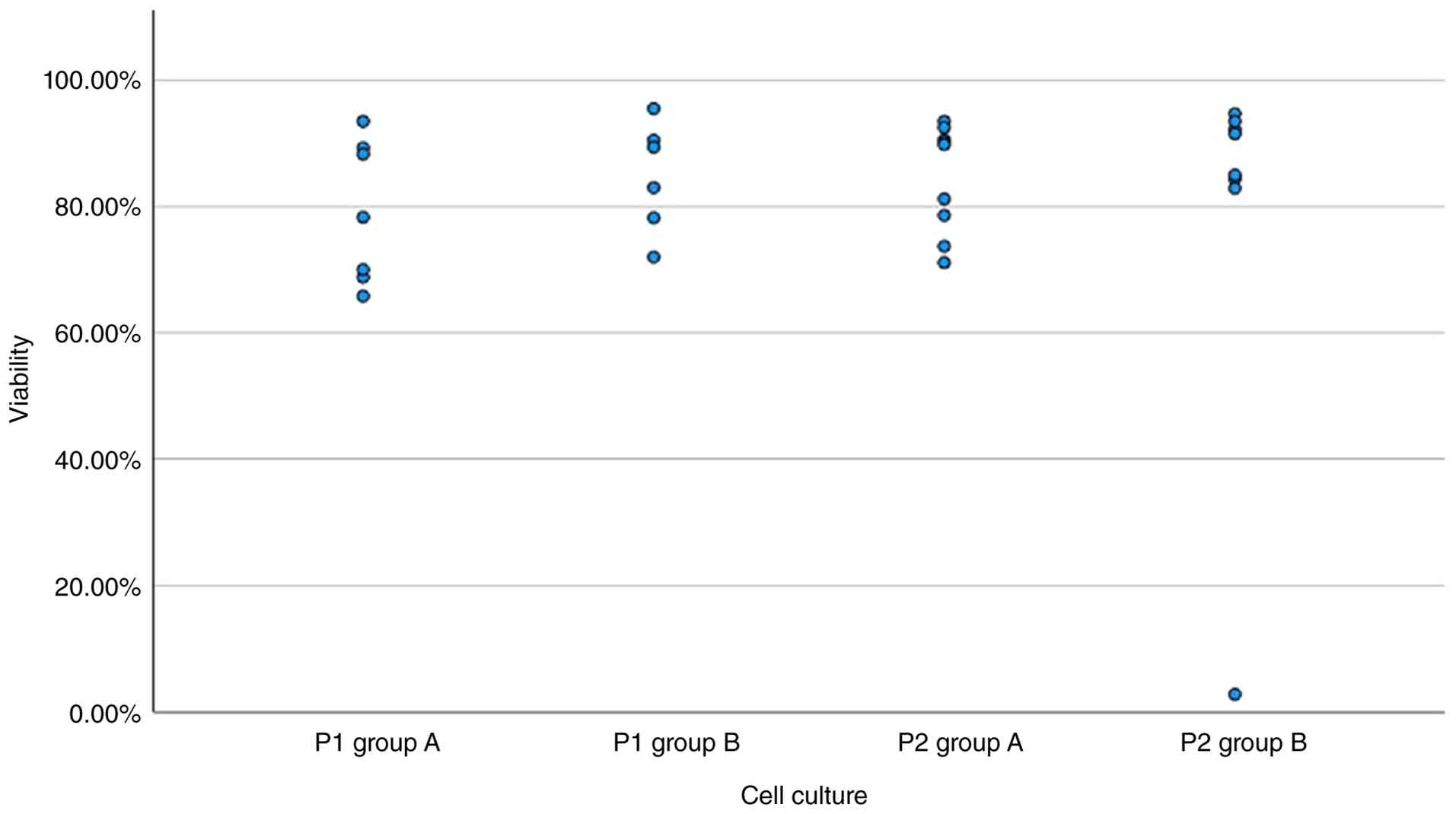
Cell viability of primary
cultures
The mean cell viability across all primary cultures
was 81.74±10.17%. The viability of culture group A was
79.14±11.27%, while the viability of group B was 84.77±8.70%. No
significant differences were observed between the two groups (group
A and B for P1; Fig. 3).
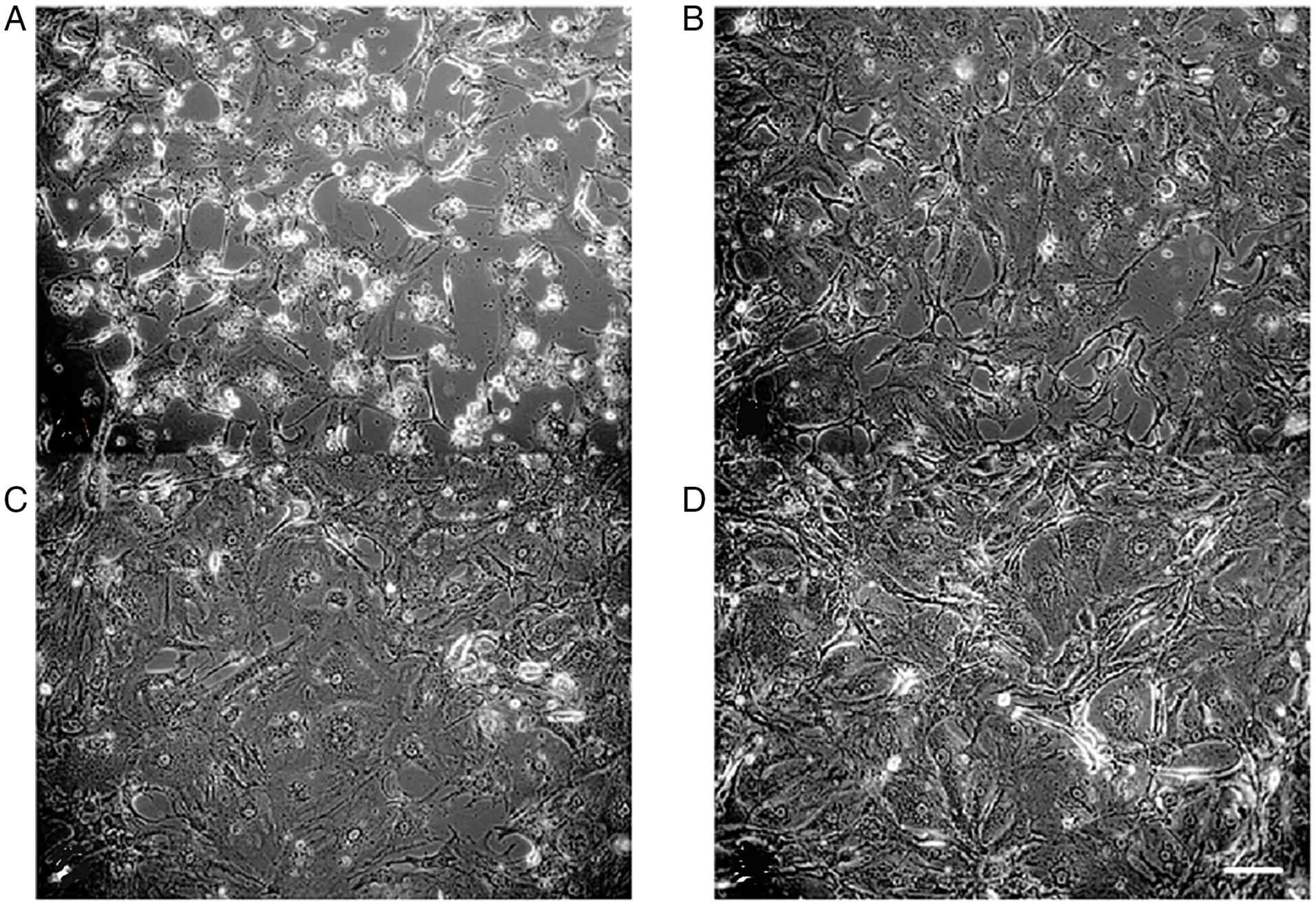
Growth rate of P2 cultures
In the secondary generation, 9 cultures from culture
A and 8 from culture B were recultivated after freezing in liquid
nitrogen for 6–7 months. Of the 17 thawed cultures, 17 were able to
continue proliferating, resulting in a re-growth rate of 94.1% for
P2. All cultures that proliferated again had established cell
attachment within 24 h. Representative cell culture images are
shown in Fig. 4.
Recultivation of culture B
When recultivating cultures that were initially
established the day after surgery, 7 out of 8 cultures (87.5%)
exhibited widespread proliferation on the culture surface in the
secondary generation (Fig. 2).
Overall proliferative performance
Comparing the two generations of cell cultures,
primary cultures showed an 85.0% cultivation success rate, whilst
secondary cultures had a success rate of 94.1%. Out of the 37
cultures derived from vital tissue or cells, 33 exhibited
widespread growth, resulting in an overall proliferative rate of
89.1% (Table II). Statistical
comparison between generations was not feasible because primary
cultures used tissue as the starting material, whereas secondary
cultures involved pre-prepared cells.
Duration until culture termination in
the secondary cell culture generation
The average time before culture termination for FISH
was 7.94±1.67 days. Culture A was terminated after an average of
7.67±1.65 days, whereas culture B was terminated after 8.25±1.75
days. The difference was minor and not statistically
significant.
Viability of secondary cultures
The average viability of all secondary cultures was
82±2%. Culture A had a viability of 85±9%, while culture B
exhibited an average viability of 78±3%. The difference observed
was not statistically significant (P=0.47; Mann-Whitney U test)
(Fig. 3). There was no evaluable
cell counting possible for culture groups A and B for samples T9077
and T9092 at P1 although there were cells detected due to technical
reasons. Therefore, there were only 7 instead of 9 tumors for group
A at P1 and 6 instead of 8 tumors for group B at P1.
LOH analysis and FISH
In four meningiomas, LOH analysis revealed no
chromosomal aberrations (Fig.
S1). A total of three tumors showed a combined loss of 1p and
22q. Additionally, an isolated 22q loss in two cases and an
isolated 1p loss in one case were observed in meningiomas (Table I).
For each meningioma, a dabbed preparation was made,
and for both culture approaches, drop preparations of both the P1
and P2 cell cultures were primed when cultures were established. In
total, up to five preparations per meningioma were analyzed using
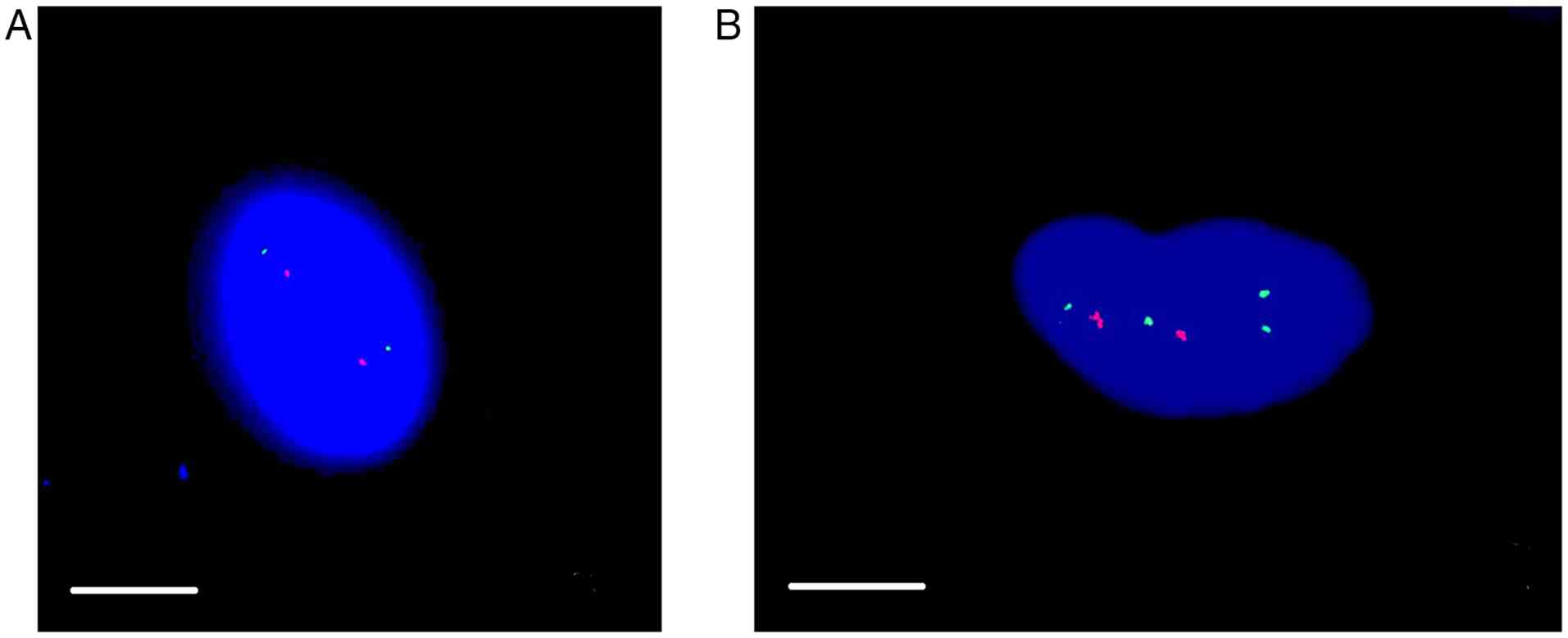
FISH. Representative images of FISH analysis are shown in Fig. 5. The results from all preparations
of a single meningioma, regardless of whether they were derived
from P1, P2 or frozen tissue, were consistent. FISH analysis
revealed an isolated loss of 22q (22q-) in three meningiomas,
combined losses of both regions 1p and 22q in three meningiomas,
and loss of 1p (1p-) in one meningioma. No chromosomal losses were
detected in three tumors.
Comparison of LOH and FISH analysis
results
FISH and LOH analysis yielded identical results in 9
out of 10 meningiomas. In 1 case, there were discrepancies: Sample
T9177 exhibited a diploid chromosome set in LOH analysis. By
contrast, FISH detected a notable loss of 22q in both drop
preparations at P1 and P2, in addition to in the dabbed
preparation.
Discussion
In the present study, meningioma tissue was
intentionally subjected to various conditions before establishing
cell cultures. The primary focus was to investigate the impact of
variations in time until further processing of the tissue
post-surgery. Other factors, such as preparation, temperature and
nutrition solution, were standardized. The present study was
performed using a well-established protocol for meningioma cell
culturing (1,9,12,20).
Specifically, the proliferative behavior of meningioma cells was
observed to assess changes when the tissue was processed for cell
culture on the day of surgery, rather than after 24-h storage of
the tissue in nutrient solution at 4°C, as was the case for the
comparison cultures. Additionally, the proliferative patterns of
secondary cultures were examined after the primary cultures were
frozen for 6–7 months and subsequently thawed. FISH analysis was
used to detect chromosomal aberrations in meningioma cells under
different conditions and preparation steps. The results from
various drop and dabbed preparations were compared with
neuropathological findings from LOH analysis. This comparison
allowed an assessment of whether the native tumor tissue was
accurately represented by the cell cultures.
Demographic factors and histopathological grading
did not influence the results (growth variability and growth rate)
in this small sample size. It was demonstrated in previous studies
(1,21–24)
that sex, WHO grading, localization of the tumor or age of the
patient did not influence meningioma cell cultures. Additionally,
clonal origin of meningioma cells could be demonstrated in cell
culture (25).
Meningioma cells were first cultured in 1978
(26). Little is known regarding
their cell behavior in meningioma cultures, despite this being a
common method in scientific research for studying tumors. The
proliferation success rate of these cells in a study reported by
Kredel (20) was 60%, and this was
observed to be 65% in another study reported by Westphal et
al (23). The proliferation
rates in the present study showed superior results compared with
the aforementioned studies. Therefore, based on these observations,
an association can be retrospectively established between the
consistency of the tumor material during sectioning for initiation
and proliferative behavior in the present study. The cell cultures
that exhibited proliferation were based on a tissue section that
predominantly had soft components and could be cut well.
In the primary generation, a proliferation rate of
90% was observed in culture A. Culture B showed sufficient
proliferation for further processing in 80% of the cultures. This
difference was not statistically significant. It can therefore be
concluded that the 24-h interval before initiating the cultures had
no real impact on the proliferative behavior of the cells.
Therefore, it is likely not necessary to process the tissue
immediately after surgery; samples could be stored in the
refrigerator and processed at a later time within the first 24 h.
In the secondary generation, all cryopreserved cultures in group A
proliferated. Of the eight frozen cultures in group B, seven were
able to be recultivated. This resulted in a proliferation rate of
87.5%. Storing cells in liquid nitrogen therefore represents a
potential method for keeping meningioma cells viable and capable of
reproduction. A direct comparison of proliferative rates between
the two cell generations was not possible due to their different
starting conditions. Primary cultures were established from native
tissue, which required cells to grow out first and then be split
after sufficient proliferation. By contrast, secondary cultures
were derived from thawed cells that only needed to proliferate in
the culture flask and were not split further. The process of cell
outgrowth from tissue, followed by proliferation and splitting at
P0 and P1, took longer compared with the proliferation of already
established cells at P2.
Another aspect to investigate would be how long
frozen meningioma cells can be cultured for and whether their
properties change during that time. Sugimoto et al (24) studied cell count, viability,
differentiation ability and aging in mesenchymal stromal cells
obtained from the pelvic bone of pediatric patients. These cells
were cryopreserved at −80°C in a commercial cryopreservation
solution with 10% DMSO and serum for a period of 1–20 years. Cell
proliferation occurred even after 20 years of cryopreservation.
However, the viability of the cells was inversely proportional to
the number of years cryopreserved; the longer the cells were
frozen, the fewer viable cells were measured in the secondary
culture. To determine possible aging of the cells, they were
examined for the deposition of senescence-associated
β-galactosidase. All cells that had been frozen for ≥5 years showed
such deposition. However, the extent of this was independent of the
number of years. This led to the conclusion that cryopreservation
does not accelerate cell aging. Furthermore, the cells in the
previous study retained their differentiation ability.
Despite largely consistent results between the LOH
findings and FISH analysis in the present study, there were
discrepancies in two tumors. These differences arose from the use
of different methods to determine the molecular cytogenetic status
of the tumor cells using PCR-based microsatellite analysis for LOH
detection or FISH. In the former, DNA was extracted from the tumor
tissue provided by the Department of Neurosurgery (Saarland
University, Hospital, Homburg, Germany), followed by PCR
amplification using primers specific for various microsatellites.
The PCR products were then separated through gel electrophoresis,
where the tumor bands were compared with the blood bands of
patients. For FISH, a different piece of tumor tissue was used to
establish cell cultures, where chromosomal alterations from these
cultured cells were analyzed using the FISH method.
To explain the different results observed in these
meningioma samples, several factors must be considered. The tissue
samples used for neuropathology and experimental neurosurgery were
obtained from different regions of the tumor, which could have
contained genetically distinct, mutated material (25). Chromosomal heterogeneity within
different regions of a meningioma has been described in previous
studies, including those by Urbschat et al (12) and Pfisterer et al (26). Additionally, the interpretation of
both methods used in the present study is examiner-dependent: In
LOH analysis, subjective judgment was involved in band evaluation
and determining chromosomal loss, whilst in FISH, counting probe
signals and determining the chromosomal number in cells also
involved subjective assessments. Nevertheless, the results from
FISH corresponded with those from LOH analysis in nine meningiomas
when considering the specific probes used.
The predominantly consistent results between the
neuropathological findings and FISH analysis led to another
insight: The cells in the cell cultures accurately represented the
original tumor tissue. Chromosomal aberrations did not change
during cell outgrowth from the tumor tissue nor after splitting the
cultures in the observed time period of the present study. This
observation has also been reported in previous studies. Lerner
et al (1) and Linsler et
al (3) described FISH as a
complement to molecular pathological examinations, enabling early
therapy for higher-grade meningiomas. It was then noted that the
results of FISH were as reliable as those of other cytogenetic
methods already established in clinical routine, where this
approach provided results in a more cost-effective and
time-efficient manner (1).
Similarly, another study by Uhlmann et al (6) used primary meningioma cell cultures
and observed an association between the tumor tissue and cultured
cells regarding cell morphology and growth characteristics.
Although not observed on a cytogenetic level, Buckley and
Eisenhardt (21) described in 1929
that meningioma cells in paraffin sections, cell cultures and vital
preparations exhibited morphological similarities.
Whilst it is acknowledged that the sample size of
the present study is limited, there are several factors to
consider. Primarily, the sample size and preparation of samples
were representative for meningiomas. Smaller sample sizes can also
yield valuable qualitative insights that are important for
understanding complex biological processes. These insights can
guide future research directions and clinical applications.
Furthermore, to the best of our knowledge, there is no literature
available providing a profound description of primary meningioma
cell culturing and growth patterns for comparison. Based on the
present data, further studies of primary cell culturing under other
conditions might be performed in the future.
In summary, the proliferative rates presented in the
present study can be utilized both within and outside of clinical
settings to estimate the success of meningioma cell culture
establishment. The present study revealed that the growth pattern
of meningioma cell cultures was independent of whether cultures
were established on the same day as surgery or 1 day post-surgery
if there were enough viable cells present in the tissue. Cell
cultures, frozen for several months or years, can be successfully
recultivated if the cells remain viable after freezing and thawing
as shown in the present study. In addition, the results of FISH
verified that cell cultures can accurately represent the tumor
material even after freezing and recultivating.
Meningioma cell culture provided a means to
accurately represent native tissue without altering the genome.
This allowed cultures to be used as a basis for investigating
tumor-specific gene mutations, similar to native tumor tissue.
Additionally, viable cells from an individual tumor were able to be
preserved for extended periods, enabling studies on tumor response
to various therapies.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Mrs. Sonja Hoffmann
(Institute of Neuropathology, Saarland University, D-66421 Homburg,
Germany) and Mrs. Sigrid Welsch (Department of Neurosurgery,
Saarland University, D-66421 Homburg, Germany) for providing
technical support.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SL and SU were responsible for conceptualizing the
present study. SL, RK, SU, JO, WSS and ALM were responsible for the
methodology. SL, WSS, JO and RK contributed towards the validation.
SL, ALM and SU performed the formal analysis. SL and SU were
responsible for investigation. SL and ALM were responsible for
writing the original draft, while SL, RK, WSS, JO, ALM and SU all
contributed towards reviewing and editing the final manuscript. SL
and ALM were also responsible for visualization, while SU and JO
provided supervision. SL, JO SU were also involved in project
administration. SU and SL confirm the authenticity of all the raw
data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
subjects involved in the present study. The present study was
approved by the Ethical Committee of the Medical Association of
Saarland (approval no. 02/20; Saarbrücken, Germany).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DMSO
|
dimethyl sulfoxide
|
|
EDTA
|
ethylenediaminetetraacetic acid
|
|
FISH
|
fluorescence in situ
hybridization
|
|
LOH
|
loss of heterozygosity
|
|
P0
|
passage 0
|
|
P1
|
passage 1
|
|
P2
|
passage 2
|
|
SSC
|
saline sodium citrate
|
References
|
1
|
Lerner C, Ketter R, Linsler S, Henn W,
Oertel J and Urbschat S: Establishment of a molecular cytogenetic
analysis for native tumor tissue of meningiomas-suitable for
clinical application. Mol Cytogenet. 7:122014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Linsler S, Ketter R, Oertel J and Urbschat
S: Fluorescence imaging of meningioma cells with somatostatin
receptor ligands: An in vitro study. Acta Neurochir (Wien).
161:1017–1024. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Linsler S, Kraemer D, Driess C, Oertel J,
Kammers K, Rahnenführer J, Ketter R and Urbschat S: Molecular
biological determinations of meningioma progression and recurrence.
PLoS One. 9:e949872014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choudhury A and Raleigh DR: Preclinical
models of meningioma: Cell culture and animal systems. Handb Clin
Neurol. 169:131–136. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ishikawa T, Matsuda M, Ishikawa H,
Toyomura J, Ohyama A, Sakamoto N, Zaboronok A and Ishikawa E:
Establishment of a novel benign meningioma cell line spontaneously
immortalized under hypoxic conditions. Hum Cell. 38:222024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Uhlmann EJ, Rabinovsky R, Varma H, El
Fatimy R, Kasper EM, Moore JM, Vega RA, Thomas AJ, Alterman RL,
Stippler M, et al: Tumor-derived cell culture model for the
investigation of meningioma biology. J Neuropathol Exp Neurol.
80:1117–1124. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jellinger K and Slowik F: Histological
subtypes and prognostic problems in meningiomas. J Neurol.
208:279–298. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ketter R, Kim YJ, Storck S, Rahnenführer
J, Romeike BF, Steudel WI, Zang KD and Henn W: Hyperdiploidy
defines a distinct cytogenetic entity of meningiomas. J Neurooncol.
83:213–221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ketter R, Rahnenführer J, Henn W, Kim YJ,
Feiden W, Steudel WI, Zang KD and Urbschat S: Correspondence of
tumor localization with tumor recurrence and cytogenetic
progression in meningiomas. Neurosurgery. 62:61–69; discussion
69–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ketter R, Urbschat S, Henn W, Feiden W,
Beerenwinkel N, Lengauer T, Steudel WI, Zang KD and Rahnenführer J:
Application of oncogenetic trees mixtures as a biostatistical model
of the clonal cytogenetic evolution of meningiomas. Int J Cancer.
121:1473–1480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sayagues JM, Tabernero MD, Maíllo A,
Trelles O, Espinosa AB, Sarasquete ME, Merino M, Rasillo A, Vera
JF, Santos-Briz A, et al: Microarray-based analysis of spinal
versus intracranial meningiomas: Different clinical, biological,
and genetic characteristics associated with distinct patterns of
gene expression. J Neuropathol Exp Neurol. 65:445–454. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Urbschat S, Rahnenführer J, Henn W, Feiden
W, Wemmert S, Linsler S, Zang KD, Oertel J and Ketter R: Clonal
cytogenetic progression within intratumorally heterogeneous
meningiomas predicts tumor recurrence. Int J Oncol. 39:1601–1608.
PubMed/NCBI
|
|
13
|
Linsler S, Müller SJ, Müller A, Senger S
and Oertel JM: Fluorescence image-guided resection of intracranial
meningioma: An experimental in vivo study on nude mice. Ann Anat.
237:1517522021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kamb A, Gruis NA, Weaver-Feldhaus J, Liu
Q, Harshman K, Tavtigian SV, Stockert E, Day RS III, Johnson BE and
Skolnick MH: A cell cycle regulator potentially involved in genesis
of many tumor types. Science. 264:436–440. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trasaktaweesakul T, Asavaritikrai P,
Talabnin K, Kongnawakul D, Tastub S, Jaturutthaweechot P, Naewwan N
and Talabnin C: In vitro properties of four benign meningioma cells
derived from WHO grade 1 meningiomas. Hum Cell. 38:842025.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van de Weijer LL, Ercolano E, Zhang T,
Shah M, Banton MC, Na J, Adams CL, Hilton D, Kurian KM and Hanemann
CO: A novel patient-derived meningioma spheroid model as a tool to
study and treat epithelial-to-mesenchymal transition (EMT) in
meningiomas. Acta Neuropathol Commun. 11:1982023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smith HL, Wadhwani N and Horbinski C:
Major features of the 2021 WHO Classification of CNS tumors.
Neurotherapeutics. 19:1691–1704. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hopman AH, Ramaekers FC, Raap AK, Beck JL,
Devilee P, van der Ploeg M and Vooijs GP: In situ hybridization as
a tool to study numerical chromosome aberrations in solid bladder
tumors. Histochemistry. 89:307–316. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hartmann C, Mueller W, Lass U, Kamel-Reid
S and von Deimling A: Molecular genetic analysis of
oligodendroglial tumors. J Neuropathol Exp Neurol. 64:10–14. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kredel FE: Tissue culture of intracranial
tumors with a note on the meningiomas. Am J Pathol. 4:337–340.
31928.PubMed/NCBI
|
|
21
|
Buckley RC and Eisenhardt L: Study of a
meningioma in supravital preparations, tissue culture and paraffin
sectios. Am J Pathol. 5:659–664. 31929.PubMed/NCBI
|
|
22
|
Kajikawa H, Kawamoto K, Herz F, Wolley RC,
Hirano A and Koss LG: Flow-through cytometry of meningiomas and
cultured meningioma cells. Acta Neuropathol. 44:183–187. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Westphal M, Hänsel M, Brunken M, König A,
Köppen JA and Herrmann HD: Initiation of primary cell cultures from
human intracranial tumors on extracellular matrix from bovine
corneal endothelial cells. Exp Cell Biol. 55:152–163.
1987.PubMed/NCBI
|
|
24
|
Sugimoto Y, Yamazaki Y, Moriyama K,
Sugimoto T, Kumazawa K, Baba K, Sone Y and Takeda A:
Differentiation and proliferation potencies of human bone
tissue-derived mesenchymal stromal cells (hBT-MSCs) after long-term
cryopreservation - Comparison among cells stored for 1, 5, 10, 15,
and 20 years. Regen Ther. 18:363–371. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
von Deimling A, Larson J, Wellenreuther R,
Stangl AP, van Velthoven V, Warnick R, Tew J Jr, Balko G and Menon
AG: Clonal origin of recurrent meningiomas. Brain Pathol.
9:645–650. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pfisterer WK, Hank NC, Preul MC, Hendricks
WP, Pueschel J, Coons SW and Scheck AC: Diagnostic and prognostic
significance of genetic regional heterogeneity in meningiomas.
Neuro Oncol. 6:290–299. 2004. View Article : Google Scholar : PubMed/NCBI
|