Introduction
Thoracic aortic aneurysm and dissection (TAFAD) is a
life-threatening condition associated with a high risk of aortic
rupture and substantial mortality. Approximately 30% of TAAD cases
demonstrate familial aggregation or carry pathogenic genetic
variants, and are collectively classified as hereditary TAAD (HTAD)
(1–3). Compared with sporadic cases, HTAD
often manifests at a younger age and may progress to dissection at
aortic diameters below conventional intervention thresholds
(4–7). These features underscore the clinical
importance of early recognition, genetic evaluation, individualized
surveillance and refined risk stratification.
Over the last decade, advances in genetic sequencing
technology and disease modeling have provided new insights into the
molecular pathogenesis and altered signaling pathways of TAAD. Core
pathogenic processes include extracellular matrix (ECM)
disorganization, dysregulated signaling networks, phenotypic and
contractile dysfunctions of vascular smooth muscle cells (vSMCs)
and altered biomechanical stress, with transforming growth factor-β
(TGF-β)-associated signaling occupying a central, but complex
position in numerous HTAD subtypes. Importantly, these mechanisms
do not stand as independent linear pathways. Rather, genetic
defects, cellular dysfunction, ECM instability and hemodynamic
forces interact dynamically within the aortic wall (8–10).
Although several recent reviews have summarized the
genetic architecture of HTAD, a comprehensive framework integrating
shared molecular pathways with subtype-specific mechanisms is still
lacking (2,11,12).
Given the marked genetic heterogeneity of HTAD, distinguishing
pathogenic mechanisms from mutation- or syndrome-specific processes
may help to explain phenotypic variability, improve
genotype-informed risk assessment and facilitate the development of
mechanism-based therapeutic strategies.
The present review therefore aims to synthesize
different strands of our current knowledge regarding the molecular
pathogenesis of HTAD, placing an emphasis on both common disease
pathways and subtype-specific features. Additionally, unresolved
controversies, gaps in the evidence and emerging research
directions were discussed, also proposing a conceptual framework
for integrating genetic findings, molecular mechanisms,
biomechanical influences and translational therapy in HTAD.
Classification of HTAD
As mentioned above, HTAD is a clinically and
genetically heterogeneous group of disorders and its classification
has important implications for diagnosis, risk stratification and
clinical management. Traditionally, HTAD has been categorized into
syndromic and non-syndromic forms, based on the presence or absence
of extra-aortic systemic manifestations (Fig. 1).
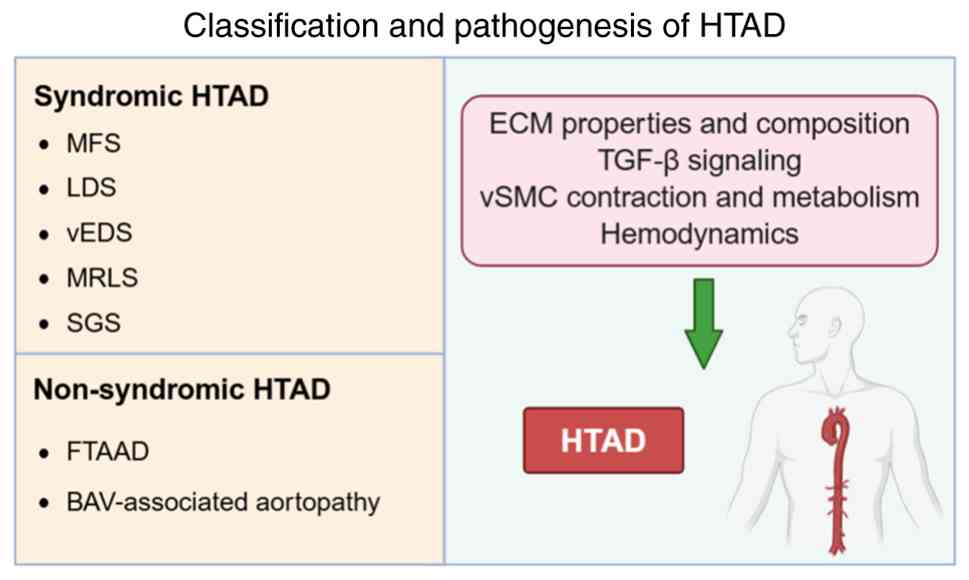 | Figure 1.Classification and pathogenesis of
HTAD. HTAD is classified into syndromic and non-syndromic forms.
These forms share common pathogenic mechanisms involving ECM
integrity and composition, dysregulation of TGF-β signaling, and
altered SMC contraction and metabolism. BAV, bicuspid aortic valve;
ECM, extracellular matrix; FTAAD, familial thoracic aortic aneurysm
and dissection; LDS, Loeys-Dietz syndrome; MFS, Marfan syndrome;
MRLS, Meester-Loeys syndrome; SGS, Shprintzen-Goldberg syndrome;
SMC, smooth muscle cell; TAAD, thoracic aortic aneurysm and
dissection; HTAD, hereditary TAAD; vEDS, vascular Ehlers-Danlos
syndrome. |
Syndromic HTAD is most commonly associated with
Mendelian connective tissue disorders and typically involves
multisystem abnormalities affecting the skin, skeleton, eyes,
craniofacial structures and vasculature (13). Major syndromic forms include Marfan
syndrome (MFS), Loeys-Dietz syndrome (LDS) and vascular
Ehlers-Danlos syndrome (vEDS). By contrast, non-syndromic HTAD is
characterized predominantly by cardiovascular involvement, and
includes conditions such as familial thoracic aortic aneurysm and
dissection (FTAAD) and bicuspid aortic valve (BAV)-associated
aortopathy (BAV is the most frequent congenital heart disease,
where the aortic valve only has two leaflets instead of the normal
three) (14).
However, this distinction is not absolute. An
increasing recognition of phenotypic overlap and convergent genetic
mechanisms has blurred the boundaries between syndromic and
non-syndromic HTAD. Taken together, these observations suggest that
HTAD is better viewed as a spectrum of associated disorders, rather
than as two entirely discrete categories.
Shared molecular mechanisms in HTAD
Despite the clinical and genetic heterogeneity of
HTAD, accumulating evidence suggests that diverse subtypes focus on
several core pathogenic mechanisms that drive aortic wall
degeneration and disease progression (Table I). These processes are highly
interconnected and collectively contribute to progressive aortic
wall degeneration.
 | Table I.Classification and causative genes of
HTAD. |
Table I.
Classification and causative genes of
HTAD.
| A, Syndromic
TAAD |
|---|
|
|---|
|
Disease/subtype | Related genes | OMIM number | Mechanism of
action |
|---|
| Marfan syndrome
signaling | FBN1 | #154700 | ECM properties and
composition; TGF-β |
| Loeys-Dietz
syndrome type I | TGFBR1 | #609192 | TGF-β
signaling |
| Loeys-Dietz
syndrome type II | TGFBR2 | #610168 |
|
| Loeys-Dietz
syndrome type III | SMAD3 | #613795 |
|
| Loeys-Dietz
syndrome type IV | TGFB2 | #614816 |
|
| Loeys-Dietz
syndrome type V | TGFB3 | #615582 |
|
| Loeys-Dietz
syndrome type VI | SMAD2 | #619656 |
|
| Loeys-Dietz
syndrome type VII | IPO8 | #605600 |
|
| Vascular
Ehlers-Danlos syndrome | COL3A1 | #130050 | ECM properties and
composition |
| Meester-Loeys
syndrome | BGN | #301870 | ECM properties and
composition |
| Shprintzen-Goldberg
syndrome | SKI | #182212 | TGF-β
signaling |
|
| B, Non-syndromic
TAAD |
|
|
Disease/subtype | Related
genes | OMIM
number | Mechanism of
action |
|
| Familial thoracic
aortic | LOX | #617168 | ECM properties and
composition |
| aneurysm
dissection | ACTA2 | #613834 | vSMC contraction
and metabolism |
|
| MYH11 | #132900 |
|
|
| MYLK | #613780 |
|
|
| PRKG1 | #615436 |
|
| Bicuspid aortic
valve | NOTCH1 | #109730 | Hemodynamics; ECM
properties and |
|
| GATA4 | #600576 | composition; vSMC
contraction |
|
| GATA5 | #611496 | and metabolism |
|
| GATA6 | #601656 |
|
|
| SMAD6 | #614823 |
|
|
| LOX | #617168 |
|
|
| ROBO4 | #618496 |
|
|
| ACTA2 | #611788 |
|
Changes in ECM properties and
composition
The aortic ECM, composed predominantly of elastin
and collagen, provides elasticity, tensile strength and structural
support to the aortic wall (15).
Disruption of ECM homeostasis weakens the aortic wall, thereby
predisposing it to aneurysm formation and dissection. In addition,
pathogenic variants in genes such as fibrillin-1 (FBN1) and
type III collagen α1 chain (COL3A1), disrupt collagen
maturation and deposition, thereby compromising the structural
integrity of the aortic wall and increasing susceptibility to
aortic disease (16,17). Although ECM dysfunction is strongly
supported by genetic and experimental evidence, the events that
link primary matrix defects to cellular and signaling abnormalities
have yet to be completely elucidated.
Dysregulation of signaling
pathways
The TGF-β signaling pathway is a key pathway that is
implicated in HTAD due to its role in vascular development, ECM
regulation and cellular homeostasis. Pathogenic variants affecting
TGF-β pathway components are associated with thoracic aortic
disease of varying severity (18–20).
In mechanistic terms, these alterations may disturb the balance
between canonical Smad-dependent signaling and non-canonical
pathways. In particular, the downstream activation of extracellular
signal-regulated kinases ½ (ERK1/2), c-Jun N-terminal kinase (JNK)
and PI3K/Akt has been implicated in maladaptive aortic remodeling
(21,22).
However, the precise contributions made by TGF-β
signaling events remains controversial, since both increased and
decreased pathway activity has been reported in the literature,
suggesting the existence of subtype-specific, stage-dependent and
compensatory effects.
Contractile and metabolic
abnormalities of vSMCs
vSMCs are essential for maintaining aortic wall
integrity, vascular tone and adaptive responses to mechanical
stress. Pathogenic variants in genes such as smooth muscle
cell-specific myosin heavy chain (MYH)11 and actin
α2, smooth muscle (ACTA2) impair the contractile function of
vSMCs and ECM synthesis, leading to a reduction in the mechanical
strength of the vascular wall (23,24).
As a result, these mutations hinder vSMCs from appropriately
sensing and responding to hemodynamic stresses, which accelerates
the degradation and dilation of the aortic wall (25–27).
In particular, increased rates of vSMC apoptosis have been
implicated in both the initiation and progression of HTAD, further
undermining aortic wall stability (28). Nevertheless, the precise
contributions made by distinct vSMC phenotypes across genetic
subtypes have yet to be fully elucidated.
Hemodynamic influences
Hemodynamic stress contributes to aortic remodeling,
particularly in BAV-associated aortopathy. Valvular abnormalities
may lead to the generation of disturbed flow, aortic regurgitation
and increased wall shear stress (WSS), all of which may promote
aortic dilation (29–31). The BAV exhibits a significant
genetic predisposition, with variants in genes including members of
the NOTCH1 and GATA families being implicated in
non-syndromic disease (32). These
variants may be predisposed not only to valve malformation, but
also to intrinsic aortic wall weakness, thereby amplifying the
effects of abnormal flow. Therefore, mechanistically speaking,
hemodynamic stress probably acts in concert with genetic
susceptibility, rather than as an isolated driver.
Considered altogether, ECM disruption, vSMC
dysfunction, dysregulated signaling and abnormal hemodynamic stress
form an interconnected pathogenic network in HTAD. Matrix defects
may alter mechanotransduction and growth factor bioavailability;
signaling abnormalities may affect vSMC phenotype and ECM
remodeling; and abnormal flow may further aggravate wall
degeneration in genetically susceptible aortas. In view of these
defects, defining the relative contribution and hierarchy of these
mechanisms will be essential in terms of improving risk prediction
and developing mechanism-based therapies.
Syndromic HTAD
MFS
MFS, an autosomal dominant connective tissue
disorder with aortic dissection represents a major life-threatening
complication. The primary causative gene, FBN1, encodes
fibrillin-1, the major structural component of extracellular
microfibrils distributed throughout multiple tissues and organs
(33). FBN1 is composed of
multiple epidermal growth factor (EGF)-like domains, the majority
of which are calcium-binding (cb)EGF-like domains. These motifs
bind extracellular calcium ions, thereby stabilizing FBN-1 and
protecting it from proteolytic degradation (34,35).
In various MFS mouse models, different degrees of
FBN1 deficiency have been observed, frequently accompanied by
aortic aneurysm formation, dissection and premature death (34). In patients with Marfan syndrome,
distinct histopathological alterations have been identified across
different affected aortic segments, which also exhibit
segment-specific patterns of ECM remodeling (36). At the molecular level, mutations in
FBN1 alter the secondary structure of EGF-like domains, leading to
protein misfolding and impaired microfibril assembly and stability.
Furthermore, structural abnormalities in cbEGF-like motifs increase
the susceptibility of FBN1 to proteolytic degradation (37). Collectively, these abnormalities in
microfibril architecture and the reduction in FBN1 levels
compromise aortic wall integrity and promote the development of
TAAD.
Beyond these structural abnormalities, studies
conducted on MFS models and patient tissues have revealed the
critical yet complex role of TGF-β in the progression of TAAD.
Accumulating evidence indicates that enhanced TGF-β signaling,
particularly through the non-canonical (Smad-independent) pathway,
is a major driver of aortic disease. Inhibition of TGF-β signaling
in myeloid cells attenuates aortic aneurysm formation in MFS mouse
models, whereas TGFB1 expression has been reported to be elevated
in patients with MFS (38,39). Studies suggest that FBN1 plays an
essential role in sequestering active TGF-β via latency-associated
peptide through its interaction with potential TGF-β binding
proteins, thereby limiting TGF-β activation (Fig. 2A) (40,41).
The loss of FBN1 integrity may therefore increase TGF-β activity,
consequently resulting in excessive TGF-β activation and
overactivation of multiple downstream non-canonical signaling
cascades, including the ERK1/2 and JNK1 pathways (Fig. 2B). Losartan, an angiotensin II
(AngII) type 1 receptor blocker, has been shown to exert protective
effects against aneurysm formation by suppressing non-canonical
ERK1/2 signaling (42).
Collectively, dysregulation of the TGF-β signaling pathway and
aberrant ERK1/2 activation are considered key contributors to
aortic aneurysm development in MFS.
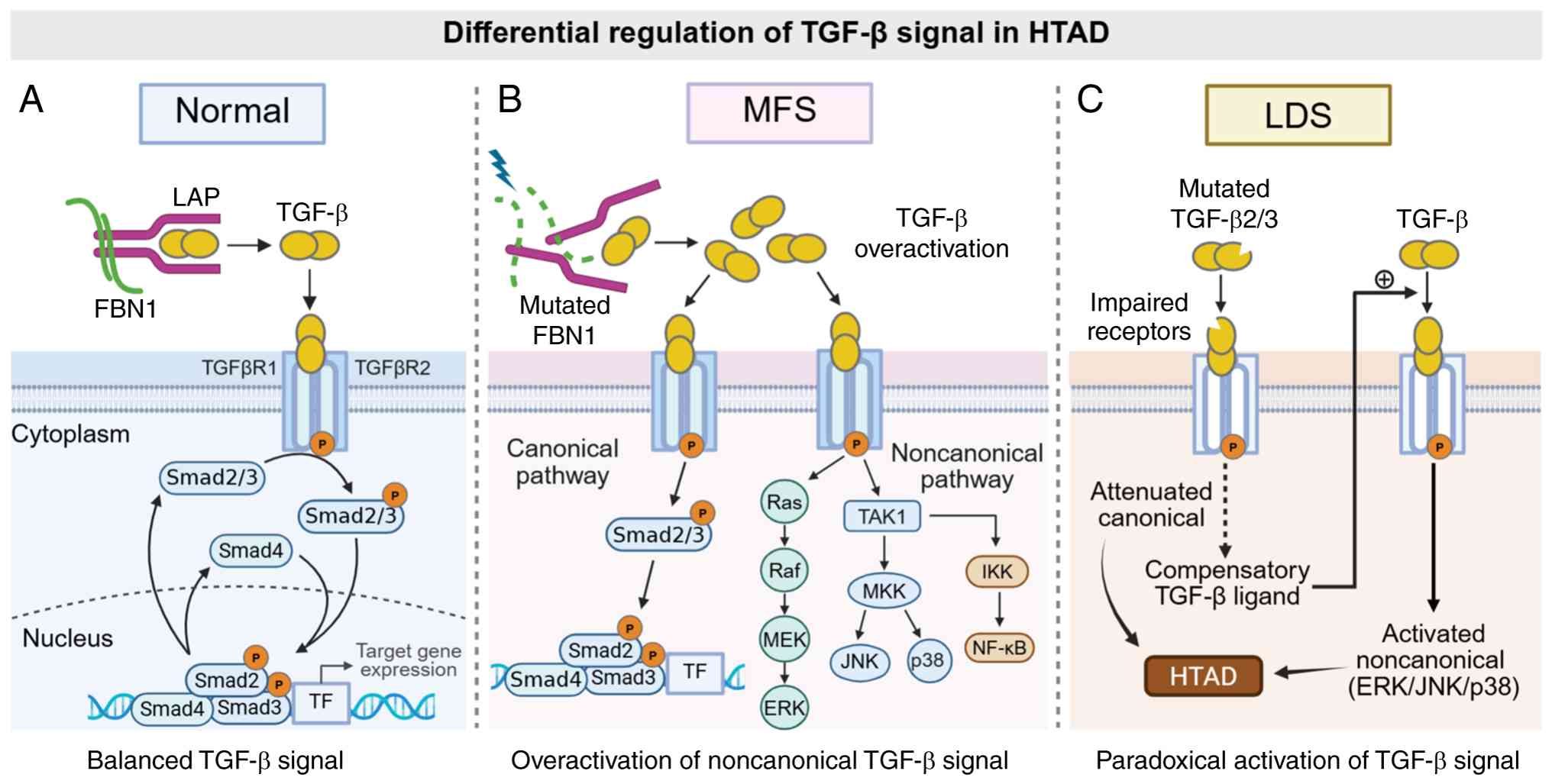 | Figure 2.Differential regulation of TGF-β
signaling in HTAD subtypes. (A) Under physiological conditions,
fibrillin-1 sequesters the latent TGF-β complex, maintaining
signaling homeostasis. (B) In MFS, pathogenic variants in FBN1 lead
to increased availability of active TGF-β and the overactivation of
noncanonical pathways, such as ERK and JNK. (C) In LDS, mutations
in TGF-β receptors impair canonical signaling; however, this
induces a compensatory upregulation of TGF-β ligands, resulting in
the paradoxical activation of downstream pathways. These mechanisms
highlight both shared and distinct features of TGF-β dysregulation
across HTAD subtypes. ERK, extracellular signal-regulated kinase;
FBN1, fibrillin-1; HTAD, hereditary thoracic aortic aneurysm and
dissection; IKK, inhibitor of nuclear factor κB kinase; JNK, c-Jun
N-terminal kinase; LAP, latency-associated peptide; LDS,
Loeys-Dietz syndrome; MEK, MAPK/ERK kinase; MFS, Marfan syndrome;
MKK, MAP kinase kinase; TAK1, transforming growth
factor-β-activated kinase 1; TF, transcription factor. |
LDS
LDS is an autosomal dominant aortic aneurysm
syndrome characterized by arterial aneurysms and tortuosity,
craniofacial abnormalities, skeletal features and cutaneous
manifestations. Although its clinical manifestations are similar to
those of MFS, typical distinguishing features of LDS include a
bifid uvula, arterial tortuosity and wide-set eyes. The current
gene-based classification acknowledges seven LDS subtypes (43). The two most common forms, LDS1 and
LDS2, are caused by pathogenic variants in TGF-β receptor 1
(TGFBR1) and TGFBR2, respectively, whereas variants
in the genes SMAD3, TGFB2, TGFB3, SMAD2 and importin
8 have also been identified in affected individuals (44–46).
Collectively, these genes encode ligands, receptors or
intracellular mediators of the TGF-β signaling pathway, thereby
rendering TGF-β dysregulation the central pathogenic feature of
LDS.
Clinical studies have consistently shown enhanced
TGF-β signaling in LDS aortic tissue across subtypes, including
increased levels of TGF-β expression, SMAD2 phosphorylation and
TGF-β target gene expression (19,46).
However, these findings were paradoxical, as the majority of the
reported TGFBR1 and TGFBR2 mutations have been shown
to reduce receptor serine/threonine kinase activity (18). Notably, increased levels of
phosphorylated SMAD2 have been observed both in LDS patient aortic
tissue with loss-of-function TGFBR1 variants and in LDS
mouse models (47,48).
Taken together, these observations suggest that
cell-autonomous defects in canonical TGF-β signaling may trigger
the compensatory upregulation of TGF-β ligand production, which, in
turn, both promotes the paracrine activation of neighboring cells
and enhances non-canonical signaling, thereby amplifying
maladaptive aortic wall remodeling (49). Therefore, LDS-associated mutations
may attenuate canonical TGF-β signaling while driving the
compensatory activation of non-canonical pathways, and the
uncovering of this mechanism helps to explain the paradoxical
increases in TGF-β expression that are observed in LDS,
underscoring the complex role of TGF-β pathway dysregulation in the
pathogenesis of HTAD (Fig.
2C).
vEDS
vEDS is one of the most severe forms of EDS and is
caused by heterozygous pathogenic variants in the gene
COL3A1. Arterial dissection or rupture, including aortic
rupture, is the leading cause of mortality in affected individuals
(50). COL3A1 encodes the
α1 chain of type III collagen. Of note, three of these chains
assemble into homotrimeric type III collagen molecules, which are
characterized by a repeating Gly-X-Y amino acid sequence. Type III
collagen is abundant in blood vessels and hollow organs, where it
provides tensile strength and tissue flexibility (51).
The majority of pathogenic COL3A1 variants in
vEDS involve glycine substitutions within the collagen
triple-helical domain, which results in the disruption of trimer
formation and the destabilization of type III collagen. In addition
to this structural mechanism, however, recent studies have
suggested that altered stress-activated signaling may modify the
severity of the disease (52–54).
In vEDS modeled mice, genetic ablation of the signaling protein
mitogen-activated protein kinase kinase 6 (Map2k6), a
p38-activating kinase, was found to increase the risk of aortic
rupture, and this was associated with reduced activation of p38 and
enhanced protein kinase C/ERK phosphorylation (53). Taken together, these findings
suggested that maladaptive crosstalk between ERK and MAP2K6/p38
signaling may exacerbate matrix-driven vascular fragility.
In conclusion, vEDS is primarily driven by
COL3A1-mediated type III collagen instability, which results in
profound arterial and visceral fragility. Emerging evidence has
demonstrated that signaling modifiers may further influence rupture
susceptibility, thereby linking ECM defects to broader
stress-response pathways in syndromic HTAD.
Non-syndromic HTAD
FTAAD
Approximately 20% of cases of non-syndromic TAAD
have exhibited familial aggregation, and this condition, which is
typically inherited in an autosomal dominant manner, is termed
FTAAD (55). Compared with
sporadic disease, FTAAD generally presents at an earlier stage and
is associated with more rapid aneurysm growth. The most significant
FTAAD-associated genes include ACTA2, MYH11, MYLK, protein
kinase cGMP-dependent 1 (PRKG1) and lysyl oxidase
(LOX) (3,56–59).
With the exception of LOX, these genes mainly affect the
vSMC contractile apparatus, leading to impaired contraction,
defective mechanosensing and phenotypic remodeling, whereas
LOX variants are primarily responsible for compromising ECM
crosslinking and biomechanical strength.
Mutations affecting the vSMC
contractile apparatus
The contractile apparatus of vSMCs consists of two
myosin heavy chains, two essential light chains and two regulatory
light chains (RLC) (25,60). Concerning the genes involved in
these processes (and as mentioned above), the SMC-specific
filament, α-actin, is encoded by ACTA2, whereas the myosin
heavy chain is encoded by MYH11. During vSMC contraction,
intracellular Ca2+ levels increase, and Ca2+
binding to calmodulin activates MYLK, which subsequently
phosphorylates RLC, resulting in the contractile force of vSMCs
(60). By contrast, during
relaxation, × cGMP-dependent protein kinase I (PKG-I), encoded by
PRKG1, activates myosin light chain phosphatase, leading to
RLC dephosphorylation and vSMC relaxation. Notably, MYLK and
PRKG1 encode key kinases involved in the regulation of
smooth muscle contraction and relaxation, respectively.
Among these genes, ACTA2 is the most common
mutation associated with FTAAD. Mutations that disrupt arginine 179
(p.R179H) and arginine 258 (p.R258C) have been shown to correlate
with aortic events (61). In the
ACTA2 knockout mouse model, aortic α-SMA expression was
reduced, accompanied by more severe aortic dilatation, which was
associated with increased levels of reactive oxygen species and
enhanced NF-κB signaling in vSMCs, as well as increased sensitivity
to exogenous AngII (62,63). Concurrently, downregulation of
α-SMA reduces the expression of integrins involved in cell-matrix
adhesion and impairs the interaction between vSMCs and the ECM,
thereby weakening aortic wall contractility (64,65).
Mutations in MYH11 are less common, but
likewise impair vSMC function. vSMC phenotypic switching from a
contractile to a synthetic state has been observed in the aortas of
patients with TAAD carrying mutations in both ACTA2 and
MYH11 (24,26,66).
This phenotypic transition is considered an important mechanism
contributing to TAAD pathogenesis. Furthermore, histological
staining of aortic sections from patients carrying MYH11
mutations revealed enhanced TGF-β signaling, which may be
associated with the pathogenesis of their TAAD. By contrast, the
upregulation of TGF-β in ACTA2 mutations exhibited a less
pronounced effect on TAAD (67).
Pathogenic variants in MYLK and PRKG1
are relatively rare; however, both have been associated with severe
TAAD by disrupting vSMC contractile function (68). Insufficient MYLK activity
reduces phosphorylation of RLC proteins, thereby impairing vSMCs'
contractile function. A recent study has further shown that
MYLK overexpression can reverse the transition of vSMC from
a contractile phenotype to a secretory phenotype, and suppress the
TGF-β signaling, ultimately attenuating TAAD progression (69). In addition, the p.Arg177Gln
mutation in PRKG1 alters the structure of its protein PKG-I,
leading to its overactivation, which results in reduced
phosphorylation of RLC in vSMC. This ultimately leads to a
reduction in vSMC contractility, thereby predisposing to aneurysm
and dissection (70).
Mutations affecting the ECM
LOX encodes a copper- dependent LOX, which
catalyzes the oxidative deamination of lysine and hydroxylysine
residues. In the ECM, LOX facilitates the formation of covalent
crosslinking between collagen and elastin, as well as the
precipitation of an insoluble matrix, making it an indispensable
component of tissue development and pathological repair (71–73).
Both missense and loss-of-function LOX variants have been
associated with extensive aortic and arterial aneurysmal disease,
which is accompanied by connective tissue manifestations (74,75).
Reduced LOX activity, along with a reduction in elastin within the
ECM, is hypothesized to impair the elasticity and tensile strength
of the aorta, thereby inducing the occurrence of TAAD.
Other ECM-associated genes affecting the ECM have
also been implicated in FTAAD, including COL3A1 and
microfibril-associated glycoprotein 5 (MFAP5), encoding a
protein involved in the interaction within FBN1 in the ECM.
Loss-of-function variants in MFAP5 may likewise represent a
potential cause for the development of FTAAD (76).
BAV-associated TAAD
A BAV is the most common congenital valvular heart
defect, with aortic stenosis and regurgitation as its most common
complications. The incidence of Stanford type A proximal aortic
dilatation TAAD is significantly increased in patients with a BAV,
indicating a strong association between valve morphology and aortic
disease (77). BAV-related TAAD
can be attributed to various factors, including genetic
predispositions, alterations in the ECM of the vascular wall and
hemodynamic changes.
The primary aortopathy hypothesis
The primary aortopathy hypothesis proposes that
BAV-associated aortic dilation arises, at least in part, as an
intrinsic developmental or genetic abnormality of the aortic wall.
NOTCH1 variants have been shown to hinder the
endothelial-mesenchymal transition of blood vessels during
embryonic development, impairing the aorta's ability to respond to
pulse pressure (29,78). GATA binding protein 4
(GATA4), GATA5 and GATA6, members of
transcription factor family, are expressed in the mesoderm during
early heart development. Variants in these genes can disrupt
transcription factor activity, leading to BAV, although their
direct contribution to TAAD remains less well defined (32). Additionally, variants in other
genes, including LOX, roundabout guidance receptor 4 and
ACTA2, may also contribute to aortic dilation through
affecting ECM integrity, endothelial stability or vSMC contractile
function.
The hemodynamic hypothesis
The hemodynamic hypothesis holds that the BAV
significantly alters the direction of blood flow and shear stress
in the ascending aorta, resulting in structural changes in the
aortic wall. Four-dimensional flow cardiovascular magnetic
resonance imaging has revealed altered blood flow patterns and
increased WSS in the aorta of patients diagnosed with a BAV
(79–81). Computational models likewise have
shown such features as a reduced valve opening area, eccentric flow
and elevated WSS compared with tricuspid aortic valves (30,82).
Collectively, these findings support a mechanistic role for
abnormal hemodynamic stress in BAV-associated aortopathy;
furthermore, a greater extent of elevated WSS has been associated
with faster aortic dilation, suggesting that WSS may serve as a
marker of disease progression (83).
However, TAAD progression may persist in certain
patients with BAV even after aortic valve replacement, suggesting
that both the primary aortopathy and hemodynamics hypotheses likely
play a role (84). Increasing
evidence suggests that their relative contribution may vary
according to the aortic segment, with aortic root dilation being
more strongly influenced by intrinsic genetic factors, and
ascending aortic dilation being more susceptible to hemodynamic
stress (85). However, definitive
separation of these effects remains challenging, since most of the
studies in the literature, to date, have lacked integrated genetic
and flow imaging data. Future studies combining genomic profiling
with advanced hemodynamic phenotyping will be essential, both to
clarify their respective roles and to improve risk stratification
in BAV-associated TAAD.
Gene-environment interactions in HTAD
Although HTAD is primarily genetically determined,
environmental and acquired factors have also been shown to modulate
disease onset and progression. Rather than acting independently,
these influences interact with the underlying genetic
susceptibility, and may contribute to variability in clinical
presentation and severity.
Hypertension is among the most established risk
factors, as chronic hypertension increases the mechanical stress on
the aortic wall, leading to damage and aneurysmal dilatation.
Patients diagnosed with HTAD are advised to regularly take
antihypertensive medications, even if their blood pressure remains
within normal limits (86).
Inflammation has been shown to facilitate the
development of aneurysmal dilatation and entrapment by disrupting
the structural integrity of the aortic wall (2,87).
In BAV-associated TAAD, elevated levels of aortic matrix
metalloproteinase (MMP)-2 and MMP-9 and increased rates of
apoptosis have been found to disrupt elastic fibers, to weaken
aortic wall strength and to promote disease progression (88–90).
However, whether inflammation is a primary driver of, or only a
secondary response to, aortic injury remains elusive.
Unhealthy lifestyles, including smoking, having a
high-fat diet and physical inactivity, may further exacerbate
disease progression of TAAD by promoting atherosclerosis and
hyperlipidemia (14). However,
evidence currently available in support of their direct role in
HTAD progression is relatively limited compared with that for
sporadic aortic disease.
Overall, these observations support a
gene-environment interaction model in HTAD in which genetic
mutations establish baseline susceptibility, whereas environmental
and acquired factors modulate disease penetrance and progression.
Future studies integrating genetic profiling with longitudinal
clinical and environmental data will be essential in order to
clarify these interactions and to improve risk stratification.
Therapeutic strategies for HTAD
ECM disruption, TGF-β dysregulation, vSMC
dysfunction and abnormal hemodynamic stress all provide potential
therapeutic targets in HTAD; however, current treatment methods
mainly aim to reduce aortic wall stress and slow disease
progression.
Current clinical pharmacotherapy
In MFS, β-blockers, including atenolol and
propranolol, remain the standard treatment, although losartan may
also be administered to slow aortic dilation in certain patients,
with the efficacy of the treatment being influenced by the FBN1
genotype (84,91).
In LDS, pharmacologic management mainly relies on
angiotensin receptor blockers and β-blockers to reduce blood
pressure and aortic wall stress (92–94).
Comparative data have suggested that losartan may be more effective
at reducing pulse wave velocity and arterial stiffness, whereas
atenolol may be more effective at lowering cardiac output and in
the treatment of stroke, supporting individualized drug selection
(95). No LDS-specific targeted
therapy has yet been approved.
Because vEDS has a distinct pathogenic basis,
therapies effective in other forms of HTAD may not be equally
beneficial. The selective β1-blocker celiprolol appears to improve
vascular integrity in vEDS animal models and is currently the
preferred preventive medication, whereas losartan has been shown to
have limited benefits in experimental models (96,97).
For FTAAD and BAV-associated TAAD, no
etiology-specific drug therapies are available at present; their
management therefore depends on antihypertensive treatment, imaging
surveillance and prophylactic surgery (98,99).
Emerging experimental strategies
Several mechanism-based approaches have shown
promise in preclinical studies. Given the central role of TGF-β
signaling in MFS and LDS, TGF-β type I receptor (TβRI/ALK5) kinase
inhibitors have been shown to be beneficial in animal models and
may represent a future targeted strategy (100,101). RNA-based therapies, including
allele-specific silencing or correction of pathogenic variants such
as MYH11, are also currently being investigated, although
efficient vascular delivery, specificity and long-term safety
remain major challenges.
Other experimental targets include inducible nitric
oxide synthase 2, the inhibition of which has also been shown to
reverse aortic dilation and medial degeneration in MFS animal
models (102). In BAV-associated
TAAD, epigenetic regulation and microRNA-based mechanisms are
currently being explored as possible therapeutic targets (103–105).
Advances that are being made in disease modeling and
biomarker development may further accelerate therapeutic discovery.
For example, patient-derived smooth muscle cell organ-on-a-chip
models can recapitulate cyclic biomechanical strain in the aortic
wall, thereby providing a platform for mechanistic studies and drug
screening (106). In parallel,
standardized biomarker validation frameworks may facilitate the
clinical application of circulating markers associated with disease
progression or treatment response (107–109).
Summary and future prospects
In the present review, HTAD has been classified into
syndromic and non-syndromic forms based on clinical phenotype.
Despite substantial genetic heterogeneity, the implicated genes
largely converge on several key processes, including ECM
homeostasis, TGF-β signaling, vSMC contraction and metabolism, and
abnormal hemodynamic stress. Across most forms of HTAD, three
shared mechanisms are prominent: ECM disruption, dysregulated TGF-β
signaling and impaired vSMC contractility.
Multiple signaling pathways have been shown to
contribute to HTAD pathogenesis. TGF-β signaling is central to
aortic homeostasis via both canonical and non-canonical pathways
(20). The PI3K/Akt signaling
pathway regulates cell survival, growth and metabolism, and has
been implicated in aortic aneurysm formation (110,111). On the other hand, NF-κB
activation promotes inflammation and ECM degradation in TAAD, and
experimental evidence suggests that targeting either PI3K/Akt or
NF-κB signaling may attenuate disease progression (112,113). Crosstalk among these pathways
further supports their potential as therapeutic targets (114).
Subtype-specific mechanisms further refine this
framework. In MFS, FBN1 mutations promote excessive
non-canonical TGF-β signaling, whereas in LDS, variants in TGF-β
receptors or SMADs reduce canonical signaling, while enhancing
compensatory non-canonical activation. In vEDS, COL3A1
mutations have been shown to primarily destabilize type III
collagen and to weaken the vascular wall, with a less prominent
role identified for TGF-β. In FTAAD, pathogenic variants mainly
affect the vSMC contractile apparatus, particularly ACTA2
and MYH11, and this is often accompanied by secondary TGF-β
activation. Finally, in BAV-associated TAAD, genetic susceptibility
interacts with abnormal wall shear stress to drive aortic
remodeling.
Overall, HTAD can be viewed as a gene-driven
disorder in which modifier genes, hemodynamic forces and
environmental factors shape disease penetrance, progression and
severity. Future studies should focus on four major priorities:
First, large genotype-phenotype registries should be set up to
improve risk stratification; secondly, patient-derived induced
pluripotent stem cells and CRISPR-engineered models should be
established to investigate disease mechanisms and therapeutic
responses; thirdly, experiments should be devised with targeted
genetic interventions, including allele-specific silencing and gene
replacement, paying particular attention to vascular delivery and
long-term safety; and fourthly and finally, polygenic and molecular
modifiers should be identified that may reveal new therapeutic
targets for both HTAD and sporadic TAAD. These efforts will help
both to refine disease prediction and to promote precision
therapies for HTAD.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82370478) and the Social Development
Project from the Key Research and Development Plan of Jiangsu
Province (grant no. BE2022731).
Availability of data and materials
Not applicable.
Authors' contributions
XW conducted the literature review and drafted the
manuscript. QT screened the articles identified through database
searches and revised the manuscript. JX organized the reviewed
literature and prepared the tables. YY contributed to the
literature review and revision of the manuscript. XT conceived and
designed the study and revised the manuscript. HH revised the
manuscript. WW edited the manuscript and supervised the project.
Data authentication is not applicable. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BAV
|
bicuspid aortic valve
|
|
ECM
|
extracellular matrix
|
|
ERK1/2
|
extracellular signal-regulated kinase
1 and 2
|
|
FTAAD
|
familial thoracic aortic aneurysm
dissection
|
|
HTAD
|
hereditary thoracic aortic aneurysm
and dissection
|
|
JNK
|
Jun N-terminal kinases
|
|
LAP
|
latency-associated peptide
|
|
LDS
|
Loeys-Dietz syndrome
|
|
LOX
|
lysyl oxidase
|
|
M2K6
|
mitogen-activated protein kinase
kinase 6
|
|
MFS
|
Marfan syndrome
|
|
MLCK
|
myosin light chain kinase
|
|
MMP
|
matrix metalloproteinase
|
|
PKG-I
|
cGMP-dependent protein kinase I
|
|
RLC
|
regulatory light chains
|
|
TAAD
|
thoracic aortic aneurysm
dissection
|
|
TF
|
transcription factors
|
|
TGF-β
|
transforming growth factor-β
|
|
vEDS
|
vascular Ehlers-Danlos syndrome
|
|
vSMCs
|
vascular smooth muscle cells
|
|
WSS
|
wall shear stress
|
References
|
1
|
Huang T and Yang B: Heritable thoracic
aortic aneurysms and dissections. Tech Vasc Interv Radiol.
24:1007472021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rodrigues Bento J, Meester J, Luyckx I,
Peeters S, Verstraeten A and Loeys B: The genetics and typical
traits of thoracic aortic aneurysm and dissection. Annu Rev
Genomics Hum Genet. 23:223–253. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wallace SE, Regalado ES, Gong L, Janda AL,
Guo DC, Russo CF, Kulmacz RJ, Hanna N, Jondeau G, Boileau C, et al:
MYLK pathogenic variants aortic disease presentation, pregnancy
risk, and characterization of pathogenic missense variants. Genet
Med. 21:144–151. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Duan Y, Xiong J, Lai Z, Zhong Y, Tian C,
Du Z, Luo Z, Yu J, Li W, Xu W, et al: Analysis of the genetic
contribution to thoracic aortic aneurysm or dissection in a
prospective cohort of patients with familial and sporadic cases in
East China. Orphanet J Rare Dis. 18:2512023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Milleron O, Arnoult F, Delorme G, Detaint
D, Pellenc Q, Raffoul R, Tchitchinadze M, Langeois M, Guien C,
Beroud C, et al: Pathogenic FBN1 genetic variation and aortic
dissection in patients with marfan syndrome. J Am Coll Cardiol.
75:843–853. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ostberg NP, Zafar MA, Ziganshin BA and
Elefteriades JA: The genetics of thoracic aortic aneurysms and
dissection: A clinical perspective. Biomolecules. 10:1822020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Isselbacher EM, Preventza O, Hamilton
Black J III, Augoustides JG, Beck AW, Bolen MA, Braverman AC, Bray
BE, Brown-Zimmerman MM, Chen EP, et al: 2022 ACC/AHA guideline for
the diagnosis and management of aortic disease: A report of the
American Heart Association/American College of Cardiology Joint
Committee on Clinical Practice Guidelines. J Am Coll Cardiol.
80:e223–e393. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chakraborty A, Li Y, Zhang C, Li Y,
Rebello KR, Li S, Xu S, Vasquez HG, Zhang L, Luo W, et al:
Epigenetic induction of smooth muscle cell phenotypic alterations
in aortic aneurysms and dissections. Circulation. 148:959–977.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng H, Min E, Baeyens N, Coon BG, Hu R,
Zhuang ZW, Chen M, Huang B, Afolabi T, Zarkada G, et al: Activation
of Smad2/3 signaling by low fluid shear stress mediates artery
inward remodeling. Proc Natl Acad Sci USA. 118:e21053391182021.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qin H, Ishiwata T, Wang R, Kudo M,
Yokoyama M, Naito Z and Asano G: Effects of extracellular matrix on
phenotype modulation and MAPK transduction of rat aortic smooth
muscle cells in vitro. Exp Mol Pathol. 69:79–90. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Isselbacher EM, Lino Cardenas CL and
Lindsay ME: Hereditary influence in thoracic aortic aneurysm and
dissection. Circulation. 133:2516–2528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rylski B, Schilling O and Czerny M: Acute
aortic dissection: Evidence, uncertainties, and future therapies.
Eur Heart J. 44:813–821. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Asta L, D'Angelo GA, Marinelli D and
Benedetto U: Genetic basis, new diagnostic approaches, and updated
therapeutic strategies of the syndromic aortic diseases: Marfan,
loeys-dietz, and vascular ehlers-danlos syndrome. Int J Environ Res
Public Health. 20:66152023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Senser EM, Misra S and Henkin S: Thoracic
aortic aneurysm: A clinical review. Cardiol Clin. 39:505–515.
2021.PubMed/NCBI
|
|
15
|
Kemberi M, Salmasi Y and Santamaria S: The
role of ADAMTS proteoglycanases in thoracic aortic disease. Int J
Mol Sci. 24:121352023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Asano K, Cantalupo A, Sedes L and Ramirez
F: Pathophysiology and therapeutics of thoracic aortic aneurysm in
marfan syndrome. Biomolecules. 12:1282022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuivaniemi H and Tromp G: Type III
collagen (COL3A1): Gene and protein structure, tissue distribution,
and associated diseases. Gene. 707:151–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Velchev JD, Van Laer L, Luyckx I, Dietz H
and Loeys B: Loeys-dietz syndrome. Adv Exp Med Biol. 1348:251–264.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schepers D, Tortora G, Morisaki H,
MacCarrick G, Lindsay M, Liang D, Mehta SG, Hague J, Verhagen J,
van de Laar I, et al: A mutation update on the LDS-associated genes
TGFB2/3 and SMAD2/3. Hum Mutat. 39:621–634. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perrotta S, Carnevale D and Lembo G: TGF-β
signalling: The Dr Jekyll and Mr Hyde of the aortic aneurysms.
Cardiovasc Res. 120:2160–2162. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang C, Chang Q, Sun X, Qian X, Liu P, Pei
H, Guo X and Liu W: Angiotensin II induces an increase in matrix
metalloproteinase 2 expression in aortic smooth muscle cells of
ascending thoracic aortic aneurysms through JNK, ERK1/2, and p38
MAPK activation. J Cardiovasc Pharmacol. 66:285–293. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Da X, Li Z, Huang X, He Z, Yu Y, Tian T,
Xu C, Yao Y and Wang QK: AGGF1 therapy inhibits thoracic aortic
aneurysms by enhancing integrin α7-mediated inhibition of TGF-β1
maturation and ERK1/2 signaling. Nat Commun. 14:22652023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo DC, Pannu H, Tran-Fadulu V, Papke CL,
Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al:
Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic
aortic aneurysms and dissections. Nat Genet. 39:1488–1493. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuang SQ, Kwartler CS, Byanova KL, Pham J,
Gong L, Prakash SK, Huang J, Kamm KE, Stull JT, Sweeney HL and
Milewicz DM: Rare, nonsynonymous variant in the smooth
muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters
force generation in the aorta and phenotype of smooth muscle cells.
Circ Res. 110:1411–1422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rombouts KB, van Merrienboer TAR, Ket JCF,
Bogunovic N, van der Velden J and Yeung KK: The role of vascular
smooth muscle cells in the development of aortic aneurysms and
dissections. Eur J Clin Invest. 52:e136972022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao G, Xuan X, Hu J, Zhang R, Jin H and
Dong H: How vascular smooth muscle cell phenotype switching
contributes to vascular disease. Cell Commun Signal. 20:1802022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen PY, Qin L, Li G, Malagon-Lopez J,
Wang Z, Bergaya S, Gujja S, Caulk AW, Murtada SI, Zhang X, et al:
Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem
Cell. 26:542–557.e11. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song W, Fu G, Li Q, Huo C, Xiao L, Liu M,
Zhang X, Sun H, Shen K, Shi L, et al: BMAL1 insufficiency increases
the risk of thoracic aortic aneurysm and dissection. Cardiovasc
Res. 122:146–161. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rashed ER, Dembar A, Riasat M and Zaidi
AN: Bicuspid aortic valves: An Up-to-date review on genetics,
natural history, and management. Curr Cardiol Rep. 24:1021–1030.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hou Q, Tao K, Du T, Wei H, Zhang H, Chen
S, Pan Y and Qiao A: A computational analysis of potential aortic
dilation induced by the hemodynamic effects of bicuspid aortic
valve phenotypes. Comput Methods Programs Biomed. 220:1068112022.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soulat G, Scott MB, Allen BD, Avery R,
Bonow RO, Malaisrie SC, McCarthy P, Fedak PWM, Barker AJ and Markl
M: Association of regional wall shear stress and progressive
ascending aorta dilation in bicuspid aortic valve. JACC Cardiovasc
Imaging. 15:33–42. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bravo-Jaimes K and Prakash SK: Genetics in
bicuspid aortic valve disease: Where are we? Prog Cardiovasc Dis.
63:398–406. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Connolly HM, Niaz T and Bowen JM: What is
marfan syndrome? JAMA. 329:16182023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zeigler SM, Sloan B and Jones JA:
Pathophysiology and Pathogenesis of Marfan Syndrome. Progress in
Heritable Soft Connective Tissue Diseases. Halper J: Springer
International Publishing; Cham: pp. 185–206. 2021, View Article : Google Scholar
|
|
35
|
Takeda N and Komuro I: Genetic basis of
hereditary thoracic aortic aneurysms and dissections. J Cardiol.
74:136–143. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Doukas P, Hruschka B, Bassett C, Buhl EM,
Simon F, Saraber P, Jacobs MJ, Uhl C, Schurgers LJ and Gombert A:
Distribution and maturity of medial collagen fibers in
thoracoabdominal Post-dissection aortic aneurysms: A comparative
study of marfan and non-marfan patients. Int J Mol Sci. 26:142024.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haller SJ, Roitberg AE and Dudley AT:
Steered molecular dynamic simulations reveal Marfan syndrome
mutations disrupt fibrillin-1 cbEGF domain mechanosensitive calcium
binding. Sci Rep. 10:168442020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dawson A, Li Y, Li Y, Ren P, Vasquez HG,
Zhang C, Rebello KR, Ageedi W, Azares AR, Mattar AB, et al:
Single-cell analysis of aneurysmal aortic tissue in patients with
marfan syndrome reveals dysfunctional TGF-β signaling. Genes.
13:952021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Holm TM, Habashi JP, Doyle JJ, Bedja D,
Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, et al:
Noncanonical TGFβ signaling contributes to aortic aneurysm
progression in Marfan syndrome mice. Science. 332:358–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lockhart-Cairns MP, Cain SA, Dajani R,
Steer R, Thomson J, Alanazi YF, Kielty CM and Baldock C: Latent
TGFβ complexes are transglutaminase cross-linked to fibrillin to
facilitate TGFβ activation. Matrix Biol. 107:24–39. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Du Q, Zhang D, Zhuang Y, Xia Q, Wen T and
Jia H: The molecular genetics of marfan syndrome. Int J Med Sci.
18:2752–2766. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Habashi JP, Judge DP, Holm TM, Cohn RD,
Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al:
Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse
model of Marfan syndrome. Science. 312:117–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Loeys BL and Dietz HC: Loeys-Dietz
Syndrome. GeneReviews(®). Adam MP, Bick S, Mirzaa GM,
Pagon RA, Wallace SE and Amemiya A: University of Washington;
Seattle: Copyright © 1993–2026, University of Washington, Seattle.
GeneReviews is a registered trademark of the University of
Washington, Seattle. All rights reserved., Seattle (WA). 1993
|
|
44
|
van de Laar IM, Oldenburg RA, Pals G,
Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM,
Willemsen R, Severijnen LA, Venselaar H, et al: Mutations in SMAD3
cause a syndromic form of aortic aneurysms and dissections with
early-onset osteoarthritis. Nat Genet. 43:121–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Boileau C, Guo DC, Hanna N, Regalado ES,
Detaint D, Gong L, Varret M, Prakash SK, Li AH, d'Indy H, et al:
TGFB2 mutations cause familial thoracic aortic aneurysms and
dissections associated with mild systemic features of Marfan
syndrome. Nat Genet. 44:916–921. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bertoli-Avella AM, Gillis E, Morisaki H,
Verhagen JMA, de Graaf BM, van de Beek G, Gallo E, Kruithof BPT,
Venselaar H, Myers LA, et al: Mutations in a TGF-β ligand, TGFB3,
cause syndromic aortic aneurysms and dissections. J Am Coll
Cardiol. 65:1324–1336. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hara H, Takeda N, Fujiwara T, Yagi H,
Maemura S, Kanaya T, Nawata K, Morita H and Komuro I: Activation of
TGF-β signaling in an aortic aneurysm in a patient with Loeys-Dietz
syndrome caused by a novel loss-of-function variant of TGFBR1. Hum
Genome Var. 6:62019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gallo EM, Loch DC, Habashi JP, Calderon
JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, et
al: Angiotensin II-dependent TGF-β signaling contributes to
Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest.
124:448–460. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bramel EE, Espinoza Camejo WA, Creamer TJ,
Restrepo L, Saqib M, Bagirzadeh R, Zeng A, Mitchell JT,
Stein-O'Brien GL, Pedroza AJ, et al: Intrinsic GATA4 expression
sensitizes the aortic root to dilation in a Loeys-Dietz syndrome
mouse model. Nat Cardiovasc Res. 3:1468–1481. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Malfait F, Castori M, Francomano CA,
Giunta C, Kosho T and Byers PH: The Ehlers-Danlos syndromes. Nat
Rev Dis Primers. 6:642020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Omar R, Malfait F and Van Agtmael T: Four
decades in the making: Collagen III and mechanisms of vascular
Ehlers Danlos Syndrome. Matrix Biol Plus. 12:1000902021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Micale L, Di Muro E, De Cegli R, Tumaini
B, Capuozzo A, Bernardi P, Morlino S, Fusco C, Nardella G, Mormone
E, et al: Multi-OMICs analysis on tridimensional fibroblast
spheroids to model vascular Ehlers-Danlos syndrome pathogenesis.
Biochim Biophys Acta Mol Basis Dis. 1871:1678962025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bowen CJ, Sorber R, Calderón Giadrosic JF,
Doyle JJ, Rykiel G, Burger Z, Zhang X, Espinoza Camejo WA, Anderson
N, Sabnis S, et al: Map2k6 is a potent genetic modifier of arterial
rupture in vascular Ehlers-Danlos syndrome mice. JCI Insight.
10:e1873152025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chiarelli N, Cinquina V, Martini P,
Bertini V, Zoppi N, Venturini M, Ritelli M and Colombi M:
Deciphering disease signatures and molecular targets in vascular
Ehlers-Danlos syndrome through transcriptome and miRNome sequencing
of dermal fibroblasts. Biochim Biophys Acta Mol Basis Dis.
1870:1669152024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Albornoz G, Coady MA, Roberts M, Davies
RR, Tranquilli M, Rizzo JA and Elefteriades JA: Familial thoracic
aortic aneurysms and Dissections-incidence, modes of inheritance,
and phenotypic patterns. Ann Thorac Surg. 82:1400–1405. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lu H, Fagnant PM, Bookwalter CS, Joel P
and Trybus KM: Vascular disease-causing mutation R258C in ACTA2
disrupts actin dynamics and interaction with myosin. Proc Natl Acad
Sci USA. 112:E4168–E4177. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Negishi K, Aizawa K, Shindo T, Suzuki T,
Sakurai T, Saito Y, Miyakawa T, Tanokura M, Kataoka Y, Maeda M, et
al: An Myh11 single lysine deletion causes aortic dissection by
reducing aortic structural integrity and contractility. Sci Rep.
12:88442022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gago-Díaz M, Blanco-Verea A, Teixidó G,
Huguet F, Gut M, Laurie S, Gut I, Carracedo Á, Evangelista A and
Brion M: PRKG1 and genetic diagnosis of early-onset thoracic aortic
disease. Eur J Clin Invest. 46:787–794. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo DC, Regalado ES, Gong L, Duan X,
Santos-Cortez RL, Arnaud P, Ren Z, Cai B, Hostetler EM, Moran R, et
al: LOX mutations predispose to thoracic aortic aneurysms and
dissections. Circ Res. 118:928–934. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fang X, Bogdanov V, Davis JP and
Kekenes-Huskey PM: Molecular insights into the MLCK Activation by
CaM. J Chem Inf Model. 63:7487–7498. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Regalado ES, Guo DC, Prakash S, Bensend
TA, Flynn K, Estrera A, Safi H, Liang D, Hyland J, Child A, et al:
Aortic disease presentation and outcome associated with ACTA2
mutations. Circ Cardiovasc Genet. 8:457–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen J, Peters A, Papke CL, Villamizar C,
Ringuette LJ, Cao J, Wang S, Ma S, Gong L, Byanova KL, et al: Loss
of smooth muscle α-Actin leads to NF-κB-dependent increased
sensitivity to Angiotensin II in smooth muscle cells and aortic
enlargement. Circ Res. 120:1903–1915. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cheng J, Zhou X, Jiang X and Sun T:
Deletion of ACTA2 in mice promotes angiotensin II induced
pathogenesis of thoracic aortic aneurysms and dissections. J Thorac
Dis. 10:4733–4740. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Massett MP, Bywaters BC, Gibbs HC,
Trzeciakowski JP, Padgham S, Chen J, Rivera G, Yeh AT, Milewicz DM
and Trache A: Loss of smooth muscle α-actin effects on
mechanosensing and cell-matrix adhesions. Exp Biol Med (Maywood).
245:374–384. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ojha KR, Kim H, Padgham S, Hopkins L,
Zamen RJ, Chattopadhyay A, Han G, Milewicz DM, Massett MP and
Trache A: Smooth Muscle-Alpha actin R149C pathogenic variant
downregulates integrin recruitment at Cell-matrix adhesions and
decreases cellular contractility. Int J Mol Sci. 24:96162023.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lu H, Du W, Ren L, Hamblin MH, Becker RC,
Chen YE and Fan Y: Vascular smooth muscle cells in aortic aneurysm:
From genetics to mechanisms. J Am Heart Assoc. 10:e0236012021.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Renard M, Callewaert B, Baetens M, Campens
L, MacDermot K, Fryns JP, Bonduelle M, Dietz HC, Gaspar IM, Cavaco
D, et al: Novel MYH11 and ACTA2 mutations reveal a role for
enhanced TGFβ signaling in FTAAD. Int J Cardiol. 165:314–321. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Regalado ES, Morris SA, Braverman AC,
Hostetler EM, De Backer J, Li R, Pyeritz RE, Yetman AT, Cervi E,
Shalhub S, et al: Comparative risks of initial aortic events
associated with genetic thoracic aortic disease. J Am Coll Cardiol.
80:857–869. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu ZL, Li Y, Lin YJ, Shi MM, Fu MX, Li
ZQ, Ning DS, Zeng XM, Liu X, Cui QH, et al: Aging aggravates aortic
aneurysm and dissection via miR-1204-MYLK signaling axis in mice.
Nat Commun. 15:59852024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Guo DC, Regalado E, Casteel DE,
Santos-Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G,
Jondeau G, et al: Recurrent gain-of-function mutation in PRKG1
causes thoracic aortic aneurysms and acute aortic dissections. Am J
Hum Genet. 93:398–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Laczko R and Csiszar K: Lysyl oxidase
(LOX): Functional contributions to signaling pathways.
Biomolecules. 10:10932020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Umana-Diaz C, Pichol-Thievend C, Marchand
MF, Atlas Y, Salza R, Malbouyres M, Barret A, Teillon J,
Ardidie-Robouant C, Ruggiero F, et al: Scavenger Receptor
cysteine-rich domains of Lysyl Oxidase-Like2 regulate endothelial
ECM and angiogenesis through non-catalytic scaffolding mechanisms.
Matrix Biol. 88:33–52. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yi X, Zhou Y, Chen Y, Feng X, Liu C, Jiang
DS, Geng J, Li X, Jiang X and Fang ZM: The expression patterns and
roles of lysyl oxidases in aortic dissection. Front Cardiovasc Med.
8:6928562021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cirnu A, Kolokotronis K, Walz K, Kilinç A,
Janz A, Williams T, Busch A, Rost S and Gerull B: Novel mutation in
LOX associates with a complex aneurysmal vascular and cardiac
phenotype. Circ Genom Precis Med. 14:e0032172021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Van Gucht I, Krebsova A, Diness BR, Laga
S, Adlam D, Kempers M, Samani NJ, Webb TR, Baranowska AA, Van Den
Heuvel L, et al: Novel LOX variants in five families with
Aortic/Arterial aneurysm and dissection with variable connective
tissue findings. Int J Mol Scie. 22:71112021. View Article : Google Scholar
|
|
76
|
Barbier M, Gross MS, Aubart M, Hanna N,
Kessler K, Guo DC, Tosolini L, Ho-Tin-Noe B, Regalado E, Varret M,
et al: MFAP5 loss-of-function mutations underscore the involvement
of matrix alteration in the pathogenesis of familial thoracic
aortic aneurysms and dissections. Am J Hum Genet. 95:736–743. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Verma R, Cohen G, Colbert J and Fedak PWM:
Bicuspid aortic valve associated aortopathy: 2022 guideline update.
Curr Opin Cardiol. 38:61–67. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Liu T, Xie M, Lv Q, Li Y, Fang L, Zhang L,
Deng W and Wang J: Bicuspid aortic valve: An update in morphology,
genetics, biomarker, complications, imaging diagnosis and
treatment. Front Physiol. 9:19212018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Rodríguez-Palomares JF, Dux-Santoy L,
Guala A, Kale R, Maldonado G, Teixidó-Turà G, Galian L, Huguet M,
Valente F, Gutiérrez L, et al: Aortic flow patterns and wall shear
stress maps by 4D-flow cardiovascular magnetic resonance in the
assessment of aortic dilatation in bicuspid aortic valve disease. J
Cardiovasc Magn Reson. 20:282018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bissell MM, Hess AT, Biasiolli L, Glaze
SJ, Loudon M, Pitcher A, Davis A, Prendergast B, Markl M, Barker
AJ, et al: Aortic dilation in bicuspid aortic valve disease: Flow
pattern is a major contributor and differs with valve fusion type.
Circ Cardiovasc Imaging. 6:499–507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Barker AJ, Markl M, Bürk J, Lorenz R, Bock
J, Bauer S, Schulz-Menger J and von Knobelsdorff-Brenkenhoff F:
Bicuspid aortic valve is associated with altered wall shear stress
in the ascending aorta. Circ Cardiovasc Imaging. 5:457–466. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sundström E and Tretter JT: Impact of
variation in commissural angle between fused leaflets in the
functionally bicuspid aortic valve on hemodynamics and tissue
biomechanics. Bioengineering (Basel). 10:12192023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tessler I, Albuisson J, Piñeiro-Sabarís R,
Verstraeten A, Kamber Kaya HE, Siguero-Álvarez M, Goudot G,
MacGrogan D, Luyckx I, Shpitzen S, et al: Novel Association of the
NOTCH pathway regulator MIB1 gene with the development of bicuspid
aortic valve. JAMA Cardiol. 8:721–731. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yang M, Nie Z, Yue H, Liang W and Wu Z:
Aortopathy associated with bicuspid aortic valve: Advances in
clinical and hemodynamics research. Front Physiol. 16:15760722025.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yassine NM, Shahram JT and Body SC:
Pathogenic mechanisms of bicuspid aortic valve aortopathy. Front
Physiol. 8:6872017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rabkin SW and Janusz MT: Aortic wall
stress in hypertension and ascending thoracic aortic aneurysms:
Implications for antihypertensive therapy. High Blood Press
Cardiovasc Prev. 20:265–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhou Z, Liu Y, Zhu X, Tang X, Wang Y, Wang
J, Xu C, Wang D, Du J and Zhou Q: Exaggerated autophagy in stanford
type A aortic dissection: A Transcriptome Pilot Analysis of Human
Ascending Aortic Tissues. Genes (Basel). 11:11872020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Haunschild J, Schellinger IN, Barnard SJ,
von Aspern K, Davierwala P, Misfeld M, Petroff D, Borger MA and Etz
CD: Bicuspid aortic valve patients show specific epigenetic tissue
signature increasing extracellular matrix destruction. Interact
Cardiovasc Thorac Surg. 29:937–943. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Balistreri CR, Pisano C, Candore G, Maresi
E, Codispoti M and Ruvolo G: Focus on the unique mechanisms
involved in thoracic aortic aneurysm formation in bicuspid aortic
valve versus tricuspid aortic valve patients: Clinical implications
of a pilot study. Eur J Cardiothorac Surg. 43:e180–e186. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chung AW, Au Yeung K, Sandor GG, Judge DP,
Dietz HC and van Breemen C: Loss of elastic fiber integrity and
reduction of vascular smooth muscle contraction resulting from the
upregulated activities of matrix metalloproteinase-2 and −9 in the
thoracic aortic aneurysm in Marfan syndrome. Circ Res. 101:512–522.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Singh MN and Lacro RV: Recent clinical
drug trials evidence in marfan syndrome and clinical implications.
Can J Cardiol. 32:66–77. 2016.PubMed/NCBI
|
|
92
|
Choo JT, Tan TH, Lai AH and Wong KY:
Loeys-Dietz syndrome: A Marfan-like syndrome associated with
aggressive vasculopathy. Singapore Med J. 50:e353–e357.
2009.PubMed/NCBI
|
|
93
|
Everitt MD, Pinto N, Hawkins JA, Mitchell
MB, Kouretas PC and Yetman AT: Cardiovascular surgery in children
with Marfan syndrome or Loeys-Dietz syndrome. J Thorac Cardiovasc
Surg. 137:1327–1333. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Sayama S, Iriyama T, Takeda N, Yamauchi H,
Toshimitsu M, Seyama T, Sone K, Kumasawa K, Nagamatsu T, Fujii T
and Osuga Y: Proposed management policy for pregnant women with
Loeys-Dietz syndrome following prophylactic aortic root replacement
based on experience from a tertiary care center. Int Heart J.
63:176–179. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sandor GG, Alghamdi MH, Raffin LA, Potts
MT, Williams LD, Potts JE, Kiess M and van Breemen C: A randomized,
double blind pilot study to assess the effects of losartan vs.
atenolol on the biophysical properties of the aorta in patients
with Marfan and Loeys-Dietz syndromes. Int J Cardiol. 179:470–475.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Dubacher N, Münger J, Gorosabel MC, Crabb
J, Ksiazek AA, Caspar SM, Bakker ENTP, van Bavel E, Ziegler U,
Carrel T, et al: Celiprolol but not losartan improves the
biomechanical integrity of the aorta in a mouse model of vascular
Ehlers-Danlos syndrome. Cardiovasc Res. 116:457–465.
2020.PubMed/NCBI
|
|
97
|
Alqahtani M, Claudinot A, Gaudry M,
Bartoli A, Barral PA, Vidal V, Boyer L, Busa T, Cadour F, Jacquier
A, et al: Endovascular management of vascular complications in
Ehlers-danlos syndrome type IV. J Clin Med. 11:63442022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Elendu C, Nzeako TR, Nwachukwu NO, Idahor
CO, Nwevo C, Bob-Ume NC and Ezeh EC: Genetic factors and management
strategies in aortic health: A literature review of inherited
aortopathy. Ann Med Surg (Lond). 87:598–615. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Atash A, Mees BME, Cramer MJ, Baas AF,
Schurgers LJ, Doevendans PA and Stillitano F: MYH11 variants in
thoracic aortic aneurysm pathophysiology: From bench to bedside.
Eur J Clin Invest. 56:e701962026. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Rhodes SD, Wu X, He Y, Chen S, Yang H,
Staser KW, Wang J, Zhang P, Jiang C, Yokota H, et al: Hyperactive
transforming growth factor-β1 signaling potentiates skeletal
defects in a neurofibromatosis type 1 mouse model. J Bone Miner
Res. 28:2476–2489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Dalal AR, Pedroza AJ, Kim JL, Gilles C, Gu
W, Kusadokoro S, Shad R, Mitchel O, Jackson W, Hiesinger W, et al:
Chemokine (C-C Motif) Ligand 2 expressing adventitial fibroblast
expansion during Loeys-Dietz syndrome aortic aneurysm formation.
Arterioscler Thromb Vasc Biol. 45:722–742. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Oller J, Méndez-Barbero N, Ruiz EJ,
Villahoz S, Renard M, Canelas LI, Briones AM, Alberca R,
Lozano-Vidal N, Hurlé MA, et al: Nitric oxide mediates aortic
disease in mice deficient in the metalloprotease Adamts1 and in a
mouse model of Marfan syndrome. Nat Med. 23:200–212. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Magouliotis DE, Sicouri S, Sicouri N,
Baudo M, Cabrucci F, Yamashita Y and Ramlawi B: Epigenetic
biomarkers in thoracic aortic aneurysm, dissection, and bicuspid
aortopathy: A comprehensive review. Biomolecules. 15:5682025.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Pasipoularides A: Clinical-pathological
correlations of BAV and the attendant thoracic aortopathies. Part
2: Pluridisciplinary perspective on their genetic and molecular
origins. J Mol Cell Cardiol. 133:233–246. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Balistreri CR, Forte M, Greco E, Paneni F,
Cavarretta E, Frati G and Sciarretta S: An overview of the
molecular mechanisms underlying development and progression of
bicuspid aortic valve disease. J Mol Cell Cardiol. 132:146–153.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Abudupataer M, Yin X, Xiang B, Chen N, Yan
S, Zhu S, Ming Y, Liu G, Zhou X, Lai H, et al: Construction of a
human aorta smooth muscle cell Organ-On-A-chip model for
recapitulating biomechanical strain in the aortic wall. J Vis Exp
Jul. 62022.doi: 10.3791/64122.
|
|
107
|
Martin-Blazquez A, Martin-Lorenzo M,
Santiago-Hernandez A, Heredero A, Donado A, Lopez JA,
Anfaiha-Sanchez M, Ruiz-Jimenez R, Esteban V, Vazquez J, et al:
Analysis of vascular smooth muscle cells from thoracic aortic
aneurysms reveals DNA damage and cell cycle arrest as hallmarks in
bicuspid aortic valve patients. J Proteome Res. 23:3012–3024. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Baratta M, Jian W, Hengel S, Kaur S,
Cunliffe J, Boer J, Hughes N, Kar S, Kellie J, Kim YJ, et al: 2023
White Paper on Recent Issues in Bioanalysis: Deuterated Drugs; LNP;
Tumor/FFPE Biopsy; Targeted Proteomics; Small Molecule Covalent
Inhibitors; Chiral Bioanalysis; Remote Regulatory Assessments;
Sample Reconciliation/Chain of Custody (PART 1A-Recommendations on
Mass Spectrometry, Chromatography, Sample Preparation Latest
Developments, Challenges, and Solutions and BMV/Regulated
Bioanalysis PART 1B-Regulatory Agencies' Inputs on Regulated
Bioanalysis/BMV, Biomarkers/IVD/CDx/BAV, Immunogenicity, Gene &
Cell Therapy and Vaccine). Bioanalysis. 16:307–364. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Fernández-Metzler C, Ackermann B, Garofolo
F, Arnold ME, DeSilva B, Gu H, Laterza O, Mao Y, Rose M,
Vazvaei-Smith F and Steenwyk R: Biomarker assay validation by mass
spectrometry. AAPS J. 24:662022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Song W, Qin L, Chen Y, Chen J and Wei L:
Single-cell transcriptome analysis identifies Versican(+)
myofibroblast as a hallmark for thoracic aortic aneurysm marked by
activation of PI3K-AKT signaling pathway. Biochem Biophys Res
Commun. 643:175–185. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zhang K, Li R, Matniyaz Y, Yu R, Pan J,
Liu W and Wang D: Liraglutide attenuates angiotensin II-induced
aortic dissection and aortic aneurysm via inhibiting M1 macrophage
polarization in APOE-/-mice. Biochem Pharmacol. 223:1161702024.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wang X and Zhang X, Qiu T, Yang Y, Li Q
and Zhang X: Dexamethasone reduces the formation of thoracic aortic
aneurysm and dissection in a murine model. Exp Cell Res.
405:1127032021. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhao K, Zhu H, He X, Du P, Liang T, Sun Y,
Jing Z and Zhou J: Senkyunolide I ameliorates thoracic aortic
aneurysm and dissection in mice via inhibiting the oxidative stress
and apoptosis of endothelial cells. Biochim Biophys Acta Mol Basis
Dis. 1869:1668192023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Luo K: Signaling cross talk between
TGF-β/Smad and other signaling pathways. Cold Spring Harb Perspect
Biol. 9:a0221372017. View Article : Google Scholar : PubMed/NCBI
|














