Introduction
A-Raf is a serine/threonine protein kinase encoded
by the RAF family. Along with BRAF and CRAF (RAF1), it is one of
the three major members of this family and serves a key role in
cell cycle and apoptosis regulation, substance transport and
metabolism, embryonic development, bone maintenance and tumor
progression (1–5). The A-Raf gene is located on band 5 of
the long arm of chromosome Xand its protein product is structurally
highly homologous to BRAF and CRAF (6,7).
Tissue expression profiles have shown that A-Raf exhibits marked
tissue specificity: A-Raf is most abundant in the epididymis,
followed by the intestine (8).
A-Raf interacts with human translocase of the outer membrane (hTOM)
on the cytoplasmic face of the mitochondrial outer membrane and
with human translocase of the inner mitochondrial membrane (hTIM)
on the inner mitochondrial membrane. hTOM and hTIM act
synergistically to mediate A-Raf transport into mitochondria, where
it is imported and anchored (9).
In mouse models, A-Raf is located in the proximal region of the
mouse X chromosome and exhibits distinct segmental expression in
the epididymis: A-Raf expression was found to be higher in the
proximal segment of the epididymis compared with the distal
segment, with the lowest level having been detected in the initial
segment; in situ hybridization results have also further
demonstrated this gradient distribution pattern (10–12).
The promoter region of the human A-Raf gene contains
three functional glucocorticoid response elements (GREs), namely
GRE-1, GRE-2 and GRE-3, which are located at −17, −34 and −168 bp,
respectively, from the transcription start site (13). The Src homology 2 (SH2) domain of
the 85 kDa regulatory subunit of PI3K can bind directly to A-Raf.
This interaction is independent of phosphorylation. The p85C-SH2
domain recognizes two distinct binding sites on A-Raf, with one
overlapping with the classical phosphotyrosine-dependent site and
the other being a phosphorylation-independent site (14). In addition, amino acids 248–267 of
A-Raf form a novel regulatory sequence referred to as the
isoform-specific hinge segment (IH-segment), which contains seven
putative phosphorylation sites. Among these, Ser257, Ser262 and
Ser264 synergistically enhance A-Raf activity, suggesting that the
IH-segment serves an important role in the precise regulation of
A-Raf function (15).
Based on research advances regarding A-Raf, the
present review summarizes its effects on the cell cycle, apoptosis
and cell proliferation, its roles in cellular transport and
metabolic processes, embryonic development and bone homeostasis,
its impact on tumor progression, the functional consequences of
genetic variants and the influence of upstream regulatory factors.
The present review therefore aimed to provide a novel perspective
for the comprehensive understanding of A-Raf functions.
A-Raf affects the cell cycle, apoptosis and
cell proliferation
With regard to cell cycle regulation, both
A-Raf-Raf-1 double knockout (DKO) and trihydrophobin1 (TH1) can
inhibit the cell cycle by modulating proliferation-associated
proteins. DKO mouse fibroblasts have exhibited delayed entry into S
phase and reduced proliferative activity, which is associated with
decreased transient phosphorylation of MEK and ERK, as well as
reduced expression levels of Fos proto-oncogene (c-Fos) and cyclin
D1 (16). This suggests that A-Raf
and Raf-1 jointly maintain normal cell cycle progression. Their
functions are complementary, such that the absence of one can be
compensated by the other, whereas simultaneous loss leads to severe
impairments in proliferation and development (16). TH1 is an endogenous negative
regulator of A-Raf that specifically binds to A-Raf and directly
inhibits its kinase activity, thereby arresting cells in the
G0/G1 phase by blocking the MAPK pathway and
downregulating c-Fos and cyclin D1 expression (3,17).
A-Raf is a key signaling hub that regulates
apoptosis (18). The subcellular
localization of A-Raf depends on the scaffolding protein kinase
suppressor of Ras 2 (KSR2). Elevated KSR2 expression promotes A-Raf
translocation to mitochondria, where it binds to and inhibits the
pro-apoptotic protein mammalian STE20-like kinase 2 (MST2), thereby
blocking apoptosis (3,18). Heterogeneous nuclear
ribonucleoprotein H (hnRNPH), acting as a transcriptional
regulator, upregulates A-Raf transcription and expression (19). A-Raf exerts an anti-apoptotic
effect by binding to and inactivating MST2 (19). Toxoplasma gondii infection,
one of the prerequisites for the inhibitory expression of A-Raf in
hosts, upregulates host microRNA-185 expression, which specifically
suppresses A-Raf expression and markedly enhances the apoptosis of
host cells (20).
A-Raf further serves a central role in the
regulation of cell proliferation. A-Raf acts in concert with CRAF
to mediate serum-induced proliferation in vascular smooth muscle
cells (21). In an
ovalbumin-induced asthma mouse model, baicalin suppressed the RAS
signaling pathway by downregulating A-Raf expression, thereby
inhibiting airway smooth muscle cell proliferation (22).
A-Raf affects cellular material transport
and metabolic processes
A-Raf influences cellular transport processes
through upstream regulation. The small G protein ARF GTPase 6
(ARF6) governs non-clathrin-mediated endocytosis and vesicular
trafficking. As a key downstream effector kinase of ARF6, A-Raf
specifically regulates the subsequent transport of endocytic
vesicles, endosomal maturation and directed transport to the
pericentriolar region (23).
A-Raf is also involved in the regulation of
carbohydrate and lipid metabolism. A-Raf specifically binds to
pyruvate kinase M2 (M2-PK) and regulates it in a
non-kinase-dependent manner, inducing a shift from a highly active
tetramer to a less active dimer, thereby inhibiting glycolysis and
promoting the Warburg effect in tumor cells (24,25).
In patients with hepatocellular carcinoma (HCC), the expression of
A-Raf and fatty acid 2-hydroxylase (FA2H) is upregulated. On the
one hand, A-Raf signaling activates the MAP2K1/ERK pathway to
promote the proliferation of HCC cells. Meanwhile, the MAP2K1/ERK
pathway upregulates FA2H expression by enhancing the expression of
the activator protein-1 (AP-1) family. Upregulated FA2H promotes
the production of 2-hydroxy fatty acid (hFA)-sphingolipids, which
further increases A-Raf expression. This positive feedback loop
accelerates HCC cell proliferation and induces increased expression
of hFA-sphingolipids ultimately promoting the progression of HCC
(Fig. 1) (26).
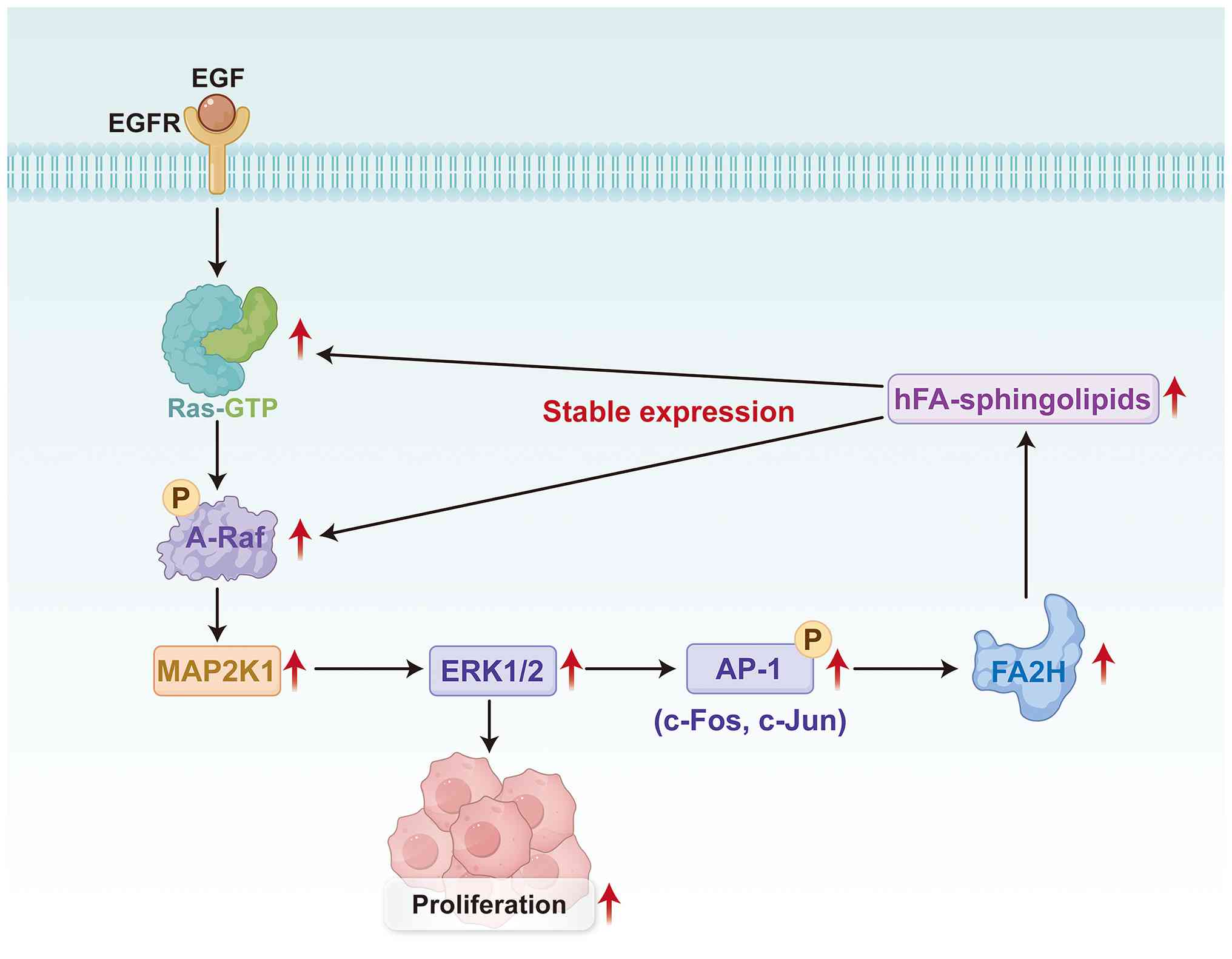 | Figure 1.Co-upregulation of A-Raf and FA2H
promotes cell proliferation and disrupts lipid metabolism in
hepatocellular carcinoma cells. Binding of EGF to EGFR induces the
conformational activation of Ras, converting it into its GTP-bound
active form. Active Ras-GTP subsequently promotes the
phosphorylation and activation of A-Raf. Activated A-Raf
phosphorylates MAP2K1, which in turn phosphorylates and activates
ERK1/2, thereby driving cell proliferation. ERK1/2 also mediates
the phosphorylation of AP-1, which subsequently upregulates FA2H.
FA2H catalyzes the biosynthesis of hFA-sphingolipids. These
hFA-sphingolipids stabilize membrane signaling complexes comprising
EGFR, Ras and A-Raf, thereby further amplifying MAP2K1 activation
and downstream signaling cascades. FA2H, fatty acid 2-hydroxylase;
hFA, 2-hydroxy fatty acid; AP-1, activator protein-1; P,
phosphorylated; c-FOS, Fos proto-oncogene. |
A-Raf serves important roles in organismal
development and bone mass maintenance
A-Raf serves a role in regulating the development of
the nervous system, the gastrointestinal tract and bones.
A-Raf-knockout mice have been shown to reach perinatal mortality
and exhibit severe defects in nervous system and gastrointestinal
tract development. These defects cannot be compensated for by other
Raf family members (B-Raf and C-Raf). This suggests that although
A-Raf deficiency does not affect embryonic development, A-Raf
serves an irreplaceable role in postnatal survival, neural
development and gastrointestinal homeostasis maintenance (5,27).
A-Raf binds to SMAD2 and catalyzes the phosphorylation of specific
sites within the SMAD2 linker region, thereby inhibiting SMAD2
nuclear translocation, attenuating its transcriptional activity,
suppressing the expression of Nodal downstream target genes and
causing endodermal defects in the embryo (28). A-Raf regulates the process of
osteogenesis by modulating the expression of
osteogenesis-associated genes (such as Runx2 and Sp7), thereby
maintaining differentiation homeostasis and contributing to bone
mass maintenance (29).
A-Raf affects tumor progression
A-Raf inhibits the progression of certain types of
lung cancer. Particularly in lung cancer types harboring wild-type
or single-mutation KRAS, elevated A-Raf expression suppresses
erb-b2 receptor tyrosine kinase 3 (ERBB3) transcription and protein
expression, thereby inhibiting lung cancer cell invasion, migration
and distant metastasis (19).
A-Raf acts as an oncogene in HCC, pancreatic cancer,
gallbladder cancer (GBC) and breast cancer (30–34).
Heterogeneous nuclear ribonucleoprotein A2 (hnRNPA2) is highly
expressed in HCC and regulates A-Raf alternative splicing by
binding to A-Raf pre-mRNA. hnRNPA2 suppresses the generation of the
dominant-negative short A-Raf isoform, upregulates full-length
A-Raf expression and enhances MAPK pathway activity, thereby
exerting oncogenic effects (30).
In addition, 5′-tRNA-derived stress-induced RNA-glutamine
downregulates A-Raf protein expression during HCC progression,
thereby inhibiting proliferation and metastasis; further suggesting
that A-Raf serves an oncogenic role in HCC (31). The long non-coding RNA linc01232 is
highly expressed in pancreatic cancer and positively regulates
A-Raf alternative splicing, resulting in increased production of
full-length functional A-Raf; thus linc01232 constitutes an
indispensable core oncogenic target in pancreatic cancer
progression and metastasis (32).
A-Raf expression is aberrantly upregulated in GBC tissues and cell
lines. Following small interfering RNA-mediated A-Raf silencing,
GBC cell proliferation, clonogenicity, migration and invasion have
been shown to be suppressed, whereas apoptosis is enhanced. These
findings indicate that A-Raf functions as an oncogene to promote
GBC progression (33). A-Raf and
RAS form protein aggregates on the cell membrane, which shield RAS
from inactivation by the tumor suppressor neurofibromin 1 (NF1) and
continuously drive breast cancer cell proliferation. Furthermore,
increased A-Raf-RAS aggregate formation confers endocrine therapy
resistance in hormone-sensitive breast cancer (34).
Effects of genetic variations on A-Raf
function
In the cytoplasm, Raf exists as a monomer.
Phosphorylation of the serine residue in the conserved region
2domain enables binding to 14-3-3 dimers, thereby maintaining a Raf
autoinhibited state. Mutations in this region disrupt 14-3-3
binding, resulting in constitutive Raf activation (35). The gain-of-function mutation
A-Raf-S214P is a causative factor in central conductive lymphatic
anomaly. Mutant A-Raf markedly upregulates ERK1/2 activity,
enhances lymphangiogenesis, disrupts the linkage between the actin
cytoskeleton and vascular endothelial cadherin, leading to abnormal
lymphatic vessel structure and leakage (36). A-Raf-S214C is a novel driver
mutation in lung adenocarcinoma; however, only 1% of patients with
lung adenocarcinoma carry A-Raf-S214 mutations, with S214C and
S214F being the most predominant variants (37).
In BRAF wild-type Langerhans cell histiocytosis, a
somatic compound mutation in A-Raf [p.Q347_A348del + p.F351L
(c.1044_1049del + c.1053C>G)] was detected within the kinase
domain. Mutant A-Raf exhibits markedly elevated MEK phosphorylation
activity compared with the wild-type form, thereby enhancing in
vitro cellular transformation and hyperactivating the MAPK/ERK
pathway (38).
A-Raf mutations are detected in 11% of intrahepatic
cholangiocarcinoma (iCCA) samples, including the A-Raf-N217I
(c.650A>T; p.Asn217Ile) and A-Raf-G322S (c.964G>A;
p.Gly322Ser) mutations. Both are heterozygous, cell-activating
mutations that are frequently observed in tumor cells and most
commonly occur in moderately to poorly differentiated
peripheral-type iCCA adenocarcinomas. By relieving autoinhibition
and enhancing dimerization, these mutations drive constitutive MAPK
pathway activation (39). The
A-Raf-R362H mutation simultaneously inhibits the dimerization of
A-Raf with itself (homodimerization) and with CRAF
(heterodimerization) and reduces the catalytic activity of A-Raf,
resulting in a kinase-deficient mutant. Compared with wild-type
A-Raf, A-Raf-R362H entirely loses its capacity to suppress ERBB3,
resulting in abrogation of the tumor-suppressive function. This
leads to hyperactivation of the ERBB3-Akt pathway in lung cancer,
markedly enhancing cellular invasion and metastasis and worsening
the clinical prognosis (Table I)
(40).
 | Table I.Mutations in A-Raf. |
Table I.
Mutations in A-Raf.
| Mutation type | Mutated site | Domain | Molecular
mechanisms | Biological
effects | (Refs.) |
|---|
| Missense
mutation | A-Raf-S214p | CR2 | Eliminates
phosphorylation sites, weakens 14-3-3 binding and lifts
self-inhibition | Primarily causes
lymphoendothelial dysplasia and lymphatic malformations | (36) |
| Missense
mutation | A-Raf-S214c | CR2 | Eliminates
phosphorylation sites, weakens 14-3-3 binding and lifts
self-inhibition | Drives the
malignant progression of lung adenocarcinoma | (37) |
| Multiplex
mutation | p.Q347_A348del +
p.F351L (c.1044_1049del + c.1053C>G) | C-terminal kinase
domain | Stabilizes the
active conformation of A-Raf and enhances its ability to form
homodimers and heterodimers with CRAF | Promotes abnormal
proliferation, differentiation and migration of Langerhans cells,
inducing malignant transformation | (38) |
| Missense
mutation | A-Raf-N217I | CR2 | Relieves the
self-inhibitory conformation of the N-terminus of A-Raf, thereby
reducing 14-3-3 binding | Promotes abnormal
proliferation and loss of cell cycle control in cholangiocarcinoma
cells, inhibits apoptosis and enhances tumor cell survival and
increases the potential for invasion and metastasis | (39) |
| Missense
mutation | A-Raf-G322S | A-Raf kinase domain
(TKD) | Locks A-Raf in an
active, open conformation to enhance its ability to recognize and
phosphorylate MEK1/2 | Promotes abnormal
proliferation and loss of cell cycle control in cholangiocarcinoma
cells, inhibits apoptosis, enhances tumor cell survival and
increases the potential for invasion and metastasis | (39) |
| Missense
mutation | A-Raf-R362H | TKD | Completely inhibits
kinase activity and lifts the transcriptional repression of
ERBB3 | Converts A-Raf from
a lung cancer metastasis inhibitor to a pro-metastasis factor,
enhancing tumor cell survival, EMT and invasive and metastatic
capabilities | (39) |
Effects of upstream regulatory factors on
A-Raf function
hnRNPH, as a splicing factor, serves an important
role in the normal expression of mature A-Raf mRNA. The oncogene
c-Myc is frequently upregulated in cancer types including
colorectal cancer, small cell lung cancer and nasopharyngeal
carcinoma, regulating the alternative splicing of A-Raf mRNA by
upregulating hnRNPH. This results in increased production of
full-length A-Raf (oncogenic) and decreased production of the
truncated form A-Rafshort (tumor-suppressive). Full-length A-Raf
binds to MST2, thereby inhibiting apoptosis and promoting cell
survival, whereas A-Rafshort lacks kinase activity and is unable to
bind to MST2. By altering the balance between these two isoforms,
c-Myc controls ERK pathway activity and influences the risk of
cellular carcinogenesis (19,41).
Upon activation of microglia by lipopolysaccharide, a marked shift
in selective polyadenylation sites of A-Raf occurs, compared with
the resting state. Alternative polyadenylation generates different
A-Raf transcript isoforms with long and short 3′ untranslated
regions. The short A-Raf mRNA isoform encodes a dominant-negative
protein lacking kinase activity, which is highly expressed in
resting microglia and limits inflammation by inhibiting the Ras-ERK
pathway (42).
Eukaryotic translation initiation factor 5A-1
(EIF5A1) is located upstream of A-Raf and markedly upregulates
A-Raf protein expression. A-Raf serves as a central signaling hub
for EIF5A1-mediated trophoblast function. By integrating
post-transcriptional mRNA regulation with the integrin/ERK
signaling pathway, A-Raf maintains the cell migration and invasion
capabilities required for placental development (43). Downregulation of A-Raf may
contribute to recurrent miscarriage (43). TH1 is a specific interacting
partner of A-Raf (44) and
selectively downregulates A-Raf. Upon binding, TH1 directly
inhibits A-Raf phosphorylation and its capacity to phosphorylate
the downstream substrate MEK. The TH1-A-Raf interaction suppresses
the expression of the downstream cell cycle-associated gene cyclin
D1, thereby reducing the proportion of cells in the S phase and
attenuating the rate of cell proliferation (17). Among the Raf family members, A-Raf
exhibits the lowest sensitivity to oncogenic Ras and tyrosine
kinases and A-Raf activation is the weakest (45).
In mouse insulinoma cells, pyruvate kinase M1
directly binds to A-Raf and upregulates its activity, which in turn
specifically activates the MEK1/ERK signaling pathway and
attenuates endoplasmic reticulum stress-induced apoptosis (46,47).
The regulatory subunit casein kinase 2 β subunit (CK2β) of the
protein kinase casein kinase 2 is a specific activator of A-Raf
capable of selectively activating A-Raf. Co-expression of CK2β and
A-Raf markedly enhances A-Raf-mediated MEK phosphorylation (by
10-fold), establishing CK2β as a potent activator of the A-Raf
pathway (48).
Treatment of cultured ventricular cardiomyocytes
with classic myocardial hypertrophy agonists, such as endothelin-1,
angiotensin II and α-adrenergic agonists, has been shown to
markedly increase A-Raf kinase activity in a time-dependent manner.
This activation in turn stimulates the downstream MEK-ERK pathway
and contributes to the regulation of pathological processes,
including cardiomyocyte hypertrophy and phenotypic remodeling
(49). IL-3 markedly upregulates
A-Raf kinase activity. This activation is entirely dependent on the
PI3K pathway. IL-3-activated A-Raf directly phosphorylates and
activates downstream MEK, thereby initiating the ERK signaling
cascade. The MEK-ERK signaling pathway is the core pathway through
which IL-3 regulates hematopoietic cell proliferation and survival.
The activation of A-Raf maintains normal proliferation and survival
of hematopoietic cells (50).
A-Raf-associated functional models
A-Raf functions as a signaling hub through a
kinase-independent pathway. There is key cross-regulation between
the RAF signaling pathway and the MST2 tumor suppressor pathway.
A-Raf inhibits MST2 by binding to and sequestering it, independent
of its own kinase activity. During epithelial cell differentiation,
the function of the A-Raf-MST2 complex is regulated by subcellular
compartmentalization. In proliferating cells of the squamous
epithelial basement membrane and in tumor cells, A-Raf localizes to
mitochondria, thereby efficiently sequestering and inhibiting MST2.
By contrast, in normal squamous epithelial cells, A-Raf is
distributed at the cell membrane, where the sequestration of MST2
is inhibited (18). A-Raf can also
upregulate and activate RAS. Upon binding to RAS, A-Raf
competitively displaces the GTPase-activating protein NF1, thereby
antagonizing NF1-mediated inhibition of RAS. This reduced
ERK-dependent inhibition of RAS and increased RAS-GTP. This
mechanism regulates the duration and downstream effects of receptor
tyrosine kinase (RTK)-induced RAS activation, where activated
RAS-GTP binds to A-Raf, induces its homo- and heterodimerization,
activates the kinase activity of A-Raf and thereby promotes cell
proliferation, sustaining signal output in RTK-dependent tumor
cells. Consequently, in human lung cancer types harboring EGFR
mutations, A-Raf amplification has been associated with acquired
resistance to EGFR inhibitors (51).
As a metabolic regulator, A-Raf has been implicated
in the maintenance of cellular energy homeostasis and the
modulation of anabolic pathways (24). A-Raf and M2-PK are interacting
proteins. A-Raf induces M2-PK dimerization, thereby inactivating
the enzyme and reducing the efficiency of glucose-to-lactate
conversion. In immortalized NIH3T3 fibroblasts, however, the
oncogenic mutant form of A-Raf increases the proportion of highly
active M2-PK tetramers, thereby enhancing the energy yield of the
glycolytic pathway (24).
A-Raf-associated treatment
A-Raf can serve as a therapeutic target to reverse
drug resistance and inhibit Ras. The highly disordered N-terminal
sequence of A-Raf drives protein self-assembly, leading to the
formation of A-Raf-Ras granule aggregates. This structure markedly
inhibits Ras GTPase-activating protein-mediated negative regulation
of NF1 membrane recruitment, thereby sustaining Ras activation.
A-Raf is a key determinant of the sensitivity of A-Raf-mutant
tumors to RAF inhibitors; knockout of A-Raf has been shown to
enhance the sensitivity of RAS-mutant cells to RAF inhibitors
(34). The use of pan-RAF
inhibitors in tumors such as non-small cell lung cancer induces
signaling reprogramming in tumor cells, activating A-Raf-mediated
compensatory bypass pathways and promoting the formation of
A-Raf-kinase suppressor of Ras 1 complexes, which sustain MAPK
signaling and contribute to the development of drug resistance.
Concurrent inhibition of RAS or MEK can effectively block this
resistance mechanism (52).
A-Raf mutations serve as predictive biomarkers for
disease diagnosis and therapeutic intervention. A patient with
advanced lung adenocarcinoma treated with oral sorafenib achieved
near-complete clinical and radiological remission sustained for up
to 5 years. Genomic and transcriptomic sequencing was performed on
primary tumor tissue samples and corresponding normal control
samples from patients. Somatic S214C mutations were detected in the
majority of tumor tissues, whereas none were identified in the
normal control samples. These findings indicate that mutated A-Raf
functions as an oncogenic driver in lung adenocarcinoma and may
serve as a predictive biomarker for sorafenib efficacy (37).
A-Raf in combination with acrylic acid-polyethylene
glycol-N-hydroxysuccinimide (AC-PEG-NHS) exerts synergistic effects
on bone repair. A-Raf mediates osteoblast differentiation induced
by mechanical stretch and is involved in osteogenesis and bone mass
maintenance. Cartilage organoids cultured with AC-PEG-NHS can
regenerate neocartilage with a gene expression profile highly
similar to that of normal healthy cartilage. Co-repair mediated by
osteoblasts and chondrocytes recapitulates the native anatomical
structure and achieves complementary mechanical properties, thereby
facilitating regulation of the bone microenvironment and shortening
of the repair cycle (53).
Challenges and prospects
As an intracellular protein kinase, A-Raf serves a
central role in the regulation of the cell cycle and apoptosis, the
transport and metabolism of substances, organismal development,
bone mass maintenance and tumor progression. Genetic variations,
such as mutations in the A-Raf gene, affect A-Raf function.
Furthermore, current research indicates that the function of A-Raf
is regulated by a number of factors, including intracellular
signaling molecules (3,18,28),
proteins involved in RNA metabolism regulation (19,20,31,41),
bioactive substances (46–50) and A-Raf-binding proteins (17,24,25,44).
However, the detailed mechanisms by which A-Raf influences the cell
cycle, apoptosis and cell proliferation require further
elucidation. However, A-Raf inhibition may promote tumor metastasis
and drug resistance through CRAF/BRAF compensatory activation, RAS
bypass or upregulation of the ERBB3-Akt pathway. Given that the
MAPK pathway regulates bone growth and the GH/IGF-1 axis, its
long-term inhibition may result in growth restriction, delayed bone
age and abnormal reproductive development, raising ethical concerns
regarding irreversible long-term hazards in pediatric patients.
Through in-depth research, it may be possible to identify patient
populations sensitive to A-Raf-targeted therapy, develop specific
inhibitors against particular A-Raf mutations and explore
combination treatment strategies that integrate A-Raf targeting
with immunotherapy, chemotherapy and radiotherapy. Considering the
important function of A-Raf in tumor progression, it is further
hypothesized that this approach not only reduces the risk of tumor
recurrence and metastasis but also improves patient prognosis,
offering novel treatment options for cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Sciences
Foundation of China (grant nos. 82571530 and 82273219), the Hunan
Provincial Natural Science Foundation (grant nos. 2025JJ50487 and
2025JJ80840), the Hunan Cancer Hospital Climb Plan (grant no.
QH2023004) and the High-Level Talent Support Program of Hunan
Cancer Hospital (grant no. 20250731-1024).
Availability of data and materials
Not applicable.
Authors' contributions
MY wrote the manuscript. MC, JW and YLi collected
the related papers and helped revise the manuscript. YZ, YLei and
CL designed and revised the manuscript. Data authentication is not
applicable. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SH2
|
Src homology 2
|
|
DKO
|
double knockout
|
|
c-FOS
|
Fos proto-oncogene
|
|
TH1
|
trihydrophobin 1
|
|
MST2
|
mammalian STE20-like kinase 2
|
|
hnRNPH
|
heterogeneous nuclear
ribonucleoprotein H
|
|
M2-PK
|
pyruvate kinase M2
|
|
HCC
|
hepatocellular carcinoma
|
|
FA2H
|
fatty acid 2-hydroxylase
|
|
GBC
|
gallbladder cancer
|
|
iCCA
|
intrahepatic cholangiocarcinoma
|
|
EIF5A1
|
eukaryotic translation initiation
factor 5A-1
|
|
CK2β
|
casein kinase 2 β subunit
|
References
|
1
|
Maurer G, Tarkowski B and Baccarini M: Raf
kinases in cancer-roles and therapeutic opportunities. Oncogene.
30:3477–3488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rebocho AP and Marais R: ARAF acts as a
scaffold to stabilize BRAF:CRAF heterodimers. Oncogene.
32:3207–3212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
An S, Yang Y, Ward R, Liu Y, Guo XX and Xu
TR: A-Raf: A new star of the family of raf kinases. Crit Rev
Biochem Mol Biol. 50:520–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galmiche A and Ezzoukhry Z: Regulation of
cell survival by RAF kinases. Med Sci (Paris). 26:729–733. 2010.(In
French). View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pritchard CA, Bolin L, Slattery R, Murray
R and McMahon M: Post-natal lethality and neurological and
gastrointestinal defects in mice with targeted disruption of the
A-Raf protein kinase gene. Curr Biol. 6:614–617. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beck TW, Huleihel M, Gunnell M, Bonner TI
and Rapp UR: The complete coding sequence of the human A-raf-1
oncogene and transforming activity of a human A-raf carrying
retrovirus. Nucleic Acids Res. 15:595–609. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ishikawa F, Takaku F, Nagao M and Sugimura
T: The complete primary structure of the rat A-raf cDNA coding
region: Conservation of the putative regulatory regions present in
rat c-raf. Oncogene Res. 1:243–253. 1987.PubMed/NCBI
|
|
8
|
Huleihel M, Goldsborough M, Cleveland J,
Gunnell M, Bonner T and Rapp UR: Characterization of murine A-raf,
a new oncogene related to the v-raf oncogene. Mol Cell Biol.
6:2655–2662. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuryev A, Ono M, Goff SA, Macaluso F and
Wennogle LP: Isoform-specific localization of A-RAF in
mitochondria. Mol Cell Biol. 20:4870–4878. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Winer MA and Wolgemuth DJ: The
segment-specific pattern of A-raf expression in the mouse
epididymis is regulated by testicular factors. Endocrinology.
136:2561–2572. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Avner P, Bućan M, Arnaud D, Lehrach H and
Rapp U: A-raf oncogene localizes on mouse X chromosome to region
some 10–17 centimorgans proximal to hypoxanthine
phosphoribosyltransferase gene. Somat Cell Mol Genet. 13:267–272.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grant SG and Chapman VM: Detailed genetic
mapping of the A-raf proto-oncogene on the mouse X chromosome.
Oncogene. 6:397–402. 1991.PubMed/NCBI
|
|
13
|
Lee JE, Beck TW, Wojnowski L and Rapp UR:
Regulation of A-raf expression. Oncogene. 12:1669–1677.
1996.PubMed/NCBI
|
|
14
|
Fang Y, Johnson LM, Mahon ES and Anderson
DH: Two phosphorylation-independent sites on the p85 SH2 domains
bind A-Raf kinase. Biochem Biophys Res Commun. 290:1267–1274. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baljuls A, Schmitz W, Mueller T, Zahedi
RP, Sickmann A, Hekman M and Rapp UR: Positive regulation of A-RAF
by phosphorylation of isoform-specific hinge segment and
identification of novel phosphorylation sites. J Biol Chem.
283:27239–27254. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mercer K, Giblett S, Oakden A, Brown J,
Marais R and Pritchard C: A-Raf and Raf-1 work together to
influence transient ERK phosphorylation and Gl/S cell cycle
progression. Oncogene. 24:5207–5217. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu W, Shen X, Yang Y, Yin X, Xie J, Yan
J, Jiang J, Liu W, Wang H, Sun M, et al: Trihydrophobin 1 is a new
negative regulator of A-Raf kinase. J Biol Chem. 279:10167–10175.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rauch J, Vandamme D, Mack B, McCann B,
Volinsky N, Blanco A, Gires O and Kolch W: Differential
localization of A-Raf regulates MST2-mediated apoptosis during
epithelial differentiation. Cell Death Differ. 23:1283–1295. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rauch J, O'Neill E, Mack B, Matthias C,
Munz M, Kolch W and Gires O: Heterogeneous nuclear
ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells
by regulating A-Raf transcription. Cancer Res. 70:1679–1688. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su D, Zhu S, Hou Z, Hao F, Xu K, Xu F, Zhu
Y, Liu D, Xu J and Tao J: Toxoplasma gondii infection
regulates apoptosis of host cells via miR-185/A-Raf axis. Parasit
Vectors. 16:3712023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cioffi CL, Garay M, Johnston JF, McGraw K,
Boggs RT, Hreniuk D and Monia BP: Selective inhibition of A-Raf and
C-Raf mRNA expression by antisense oligodeoxynucleotides in rat
vascular smooth muscle cells: Role of A-Raf and C-Raf in
serum-induced proliferation. Mol Pharmacol. 51:383–389. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu L, Li L, Yan C, Cao Y, Duan X and Sun
J: Baicalin inhibits airway smooth muscle cells proliferation
through the ras signaling pathway in murine asthmatic airway
remodeling model. Oxid Med Cell Longev. 2023:41441382023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nekhoroshkova E, Albert S, Becker M and
Rapp UR: A-RAF kinase functions in ARF6 regulated endocytic
membrane traffic. PLoS One. 4:e46472009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mazurek S, Drexler HCA, Troppmair J,
Eigenbrodt E and Rapp UR: Regulation of pyruvate kinase type M2 by
A-Raf: A possible glycolytic stop or go mechanism. Anticancer Res.
27:3963–3971. 2007.PubMed/NCBI
|
|
25
|
Mazurek S, Boschek CB, Hugo F and
Eigenbrodt E: Pyruvate kinase type M2 and its role in tumor growth
and spreading. Semin Cancer Biol. 15:300–308. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ranjpour M, Wajid S and Jain SK: Elevated
expression of A-Raf and FA2H in hepatocellular carcinoma is
associated with lipid metabolism dysregulation and cancer
progression. Anticancer Agents Med Chem. 19:236–247. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Udell CM, Rajakulendran T, Sicheri F and
Therrien M: Mechanistic principles of RAF kinase signaling. Cell
Mol Life Sci. 68:553–565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu X, Xiong C, Jia S, Zhang Y, Chen YG,
Wang Q and Meng A: Araf kinase antagonizes Nodal-Smad2 activity in
mesendoderm development by directly phosphorylating the Smad2
linker region. Nat Commun. 4:17282013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Q, Matsui H, Horiuchi H, Liang X and
Sasaki K: A-Raf and C-Raf differentially regulate mechanobiological
response of osteoblasts to guide mechanical stress-induced
differentiation. Biochem Biophys Res Commun. 476:438–444. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shilo A, Ben Hur V, Denichenko P, Stein I,
Pikarsky E, Rauch J, Kolch W, Zender L and Karni R: Splicing factor
hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf
splicing in hepatocellular carcinoma development. RNA. 20:505–515.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu C, Liu D, Zhang L, Wang J, Ding Y, Sun
Z and Wang W: 5′-tiRNA-Gln inhibits hepatocellular carcinoma
progression by repressing translation through the interaction with
eukaryotic initiation factor 4A-I. Front Med. 17:476–492. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng LD, Shi GD, Ge WL, Huang XM, Chen Q,
Yuan H, Wu PF, Lu YC, Shen P, Zhang YH, et al: Linc01232 promotes
the metastasis of pancreatic cancer by suppressing the
ubiquitin-mediated degradation of HNRNPA2B1 and activating the
A-Raf-induced MAPK/ERK signaling pathway. Cancer Lett. 494:107–120.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin W, Tong C, Zhang W, Cen W, Wang Y, Li
J, Zhu Z, Yu J and Lu B: Silencing ARAF suppresses the malignant
phenotypes of gallbladder cancer cells. Biomed Res Int.
2020:32357862020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li W, Shi X, Tan C, Jiang Z, Li M, Ji Z,
Zhou J, Luo M, Fan Z, Ding Z, et al: Plasma membrane-associated
ARAF condensates fuel RAS-related cancer drug resistance. Nat Chem
Biol. 21:1226–1237. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lavoie H and Therrien M: Regulation of RAF
protein kinases in ERK signalling. Nat Rev Mol Cell Biol.
16:281–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li D, March ME, Gutierrez-Uzquiza A, Kao
C, Seiler C, Pinto E, Matsuoka LS, Battig MR, Bhoj EJ, Wenger TL,
et al: ARAF recurrent mutation causes central conducting lymphatic
anomaly treatable with a MEK inhibitor. Nat Med. 25:1116–1122.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Imielinski M, Greulich H, Kaplan B, Araujo
L, Amann J, Horn L, Schiller J, Villalona-Calero MA, Meyerson M and
Carbone DP: Oncogenic and sorafenib-sensitive ARAF mutations in
lung adenocarcinoma. J Clin Invest. 124:1582–1586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nelson DS, Quispel W, Badalian-Very G, van
Halteren AG, van den Bos C, Bovée JV, Tian SY, Van Hummelen P,
Ducar M, MacConaill LE, et al: Somatic activating ARAF mutations in
Langerhans cell histiocytosis. Blood. 123:3152–1355. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sia D, Losic B, Moeini A, Cabellos L, Hao
K, Revill K, Bonal D, Miltiadous O, Zhang Z, Hoshida Y, et al:
Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion
and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun.
6:60872015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mooz J, Riegel K, Ps H, Sadanandam A,
Marini F, Klein M, Werner U, Roth W, Wilken-Schmitz A, Tegeder I
and Rajalingam K: ARAF suppresses ERBB3 expression and metastasis
in a subset of lung cancers. Sci Adv. 8:eabk15382022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rauch J, Moran-Jones K, Albrecht V,
Schwarzl T, Hunter K, Gires O and Kolch W: c-Myc regulates RNA
splicing of the A-Raf kinase and its activation of the ERK pathway.
Cancer Res. 71:4664–4674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hwang HW, Saito Y, Park CY, Blachère NE,
Tajima Y, Fak JJ, Zucker-Scharff I and Darnell RB: cTag-PAPERCLIP
reveals alternative polyadenylation promotes cell-type specific
protein diversity and shifts Araf isoforms with microglia
activation. Neuron. 95:1334–1349.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang J, Mo HQ, Tian FJ, Zeng WH, Liu XR,
Ma XL, Li X, Qin S, Fan CF and Lin Y: EIF5A1 promotes trophoblast
migration and invasion via ARAF-mediated activation of the
integrin/ERK signaling pathway. Cell Death Dis. 9:9262018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yin XL, Chen S and Gu JX: Identification
of TH1 as an interaction partner of A-Raf kinase. Mol Cell Biochem.
231:69–74. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marais R, Light Y, Paterson HF, Mason CS
and Marshall CJ: Differential regulation of Raf-1, A-Raf, and B-Raf
by oncogenic ras and tyrosine kinases. J Biol Chem. 272:4378–4383.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu X, Noh SJ, Zhou G, Dixon JE and Guan
KL: Selective activation of MEK1 but not MEK2 by A-Raf from
epidermal growth factor-stimulated Hela cells. J Biol Chem.
271:3265–3271. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Horiuchi Y, Nakatsu D, Kano F and Murata
M: Pyruvate kinase M1 interacts with A-Raf and inhibits endoplasmic
reticulum stress-induced apoptosis by activating MEK1/ERK pathway
in mouse insulinoma cells. Cell Signal. 38:212–222. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hagemann C, Kalmes A, Wixler V, Wixler L,
Schuster T and Rapp UR: The regulatory subunit of protein kinase
CK2 is a specific A-Raf activator. FEBS Lett. 403:200–202. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bogoyevitch MA, Marshall CJ and Sugden PH:
Hypertrophic agonists stimulate the activities of the protein
kinases c-Raf and A-Raf in cultured ventricular myocytes. J Biol
Chem. 270:26303–26310. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sutor SL, Vroman BT, Armstrong EA, Abraham
RT and Karnitz LM: A phosphatidylinositol 3-kinase-dependent
pathway that differentially regulates c-Raf and A-Raf. J Biol Chem.
274:7002–7010. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Su W, Mukherjee R, Yaeger R, Son J, Xu J,
Na N, Merna Timaul N, Hechtman J, Paroder V, Lin M, et al: ARAF
protein kinase activates RAS by antagonizing its binding to RASGAP
NF1. Mol Cell. 82:2443–2457.e7. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Manabe T, Bergo HC, Li Q, Wang TS,
Severson P, Miller N, Lee C, Yay Donderici E, Zhang N, Wu W, et al:
Pan-RAF inhibitor exarafenib targets BRAF class II/III NSCLC and
reveals ARAF-KSR1 resistance and combination strategies. Nat
Commun. 17:24842026. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shen CY, Zhou QR, Wu X, Han XY, Zhang Q,
Chen X, Lai YX, Bai L, Jing YY, Wang JH, et al: Accelerating
cartilage regeneration with DNA-SF hydrogel sustained release
system-based cartilage organoids. Mil Med Res. 12:392025.PubMed/NCBI
|














