Introduction
Malignant glioma is a highly aggressive tumor with a
poor prognosis despite advances in surgery, radiotherapy and
chemotherapy (1). Temozolomide
(TMZ), a new generation of oral alkylating agents that induces
apoptosis by methylation of the O6-position of guanine,
is considered to be one of the most effective chemotherapeutic
agents against malignant glioma (2). Its clinical efficacy is limited in
most cases by recurrence or the development of resistance (3). Various approaches have been
investigated to overcome the resistance and improve the efficacy of
TMZ, including dose-intense scheduling (4), combined therapy with radiotherapy
(2), angiogenesis inhibition
(5), immune modulators (6) and other chemotherapies (7). The mechanism of TMZ resistance remains
unclear. An understanding of the resistance mechanism is crucial if
the clinical efficacy of TMZ is to be improved.
Signal transducers and activators of transcription 3
(STAT3) is a cytoplasmic transcription factor that is
phosphorylated by various protein tyrosine kinases including Janus
kinases and Src in response to the activation of cytokines and
growth factors (8,9). Phosphorylation at tyrosine 705
(pSTAT3-Tyr705) induces its dimerization, nuclear translocation and
DNA binding (10), and
phosphorylation at Serine 727 (pSTAT3-Ser727) via the MAPK or mTOR
pathway appears to regulate transcriptional activation (8,11).
STAT3 plays a key role in cell proliferation, apoptosis and
differentiation (12). STAT3 is
transiently phosphorylated in normal cells, whereas it is
constitutively activated in the majority of solid tumor cells
(13). Aberrant activation of this
protein affects tumorigenesis and interferes with tumor
chemotherapy (14). The results of
numerous studies are in agreement with the suggestion that the
STAT3 oncogenic pathway is related to drug resistance and the
prevention of cell death through intrinsic and/or extrinsic
apoptotic pathways. Constitutively-activated STAT3 up-regulates the
expression of prosurvival proteins, including Bcl-xL and Mcl-1,
which prevent cytochrome c release and inhibit
mitochondria-dependent cell death (15). In addition, STAT3 has been reported
to exhibits resistance to Fas-mediated apoptosis in multiple
myeloma (16). Activated STAT3 is
overexpressed in the majority of paclitaxel-resistant ovarian
cancer cells, and therefore the inhibition of STAT3 activation
results in a significantly decreased paclitaxel resistance and
enhanced apoptosis (17).
Drug-resistant recurrent tumors have increased STAT3
phosphorylation as compared with matched primary tumors.
This study was therefore designed to investigate the
relationship between STAT3 and TMZ-resistance in gliomas.
Materials and methods
Cell lines and culture conditions
Human glioma cell lines U373, U251 and T98G were
purchased from the American Tissue Culture Collection (Rockville,
MD, USA). To develop TR cells, U373, U251 and T98G cell lines were
initially cultured in the presence of 12.5 μM of TMZ. The
concentration was then increased by 2-fold for every two passages
until it reached 800 μM (maximum concentration). The cancer cell
lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM;
WelGene, Dae-Ku, Korea) supplemented with 10% fetal bovine serum
(FBS; WelGene) and 50 μg/ml gentamicin (Gibco, Grand Island, NY,
USA).
Drugs
Stock solutions of TMZ (Schering-Plough, Kenilworth,
NJ, USA) and Celecoxib (Pfizer, New York, NY, USA) were prepared by
dissolving the drug in dimethyl sulfoxide (Sigma-Aldrich, St.
Louis, MO, USA). Methotrexate (Sigma-Aldrich) was prepared by
dissolving the drug in 10 mM sodium chloride.
1,3-Bis(2-chloroethyl)-1-nitrosourea (BCNU; Bristol Myers Squibb,
Princeton, NJ, USA) was reconstituted in sterile water.
Cell viability assay
Cell viability was assessed using the Cell Counting
Kit-8 assay (CCK-8; Dojindo, Japan). Briefly, 500 cells (U373,
U251, TR-U373 and TR-U251) were plated in each well of a 96-well
plate. After 24 h, the cells were treated with various
concentrations of drugs including TMZ (20 mg/ml), BCNU (3.3 mg/ml),
MTX (1.5 μg/ml) and Celecoxib (100 mg/ml) for 5 days and the
dose-response was analyzed. The cells were incubated with CCK-8
solution for 2 h and the absorbance was measured at 450 nm using an
ELISA reader (Molecular Devices, Sunnyvale, CA, USA). The
percentage of cell viability was calculated relative to the
untreated cells as the control.
Apoptosis analysis by flow cytometry
An Annexin V-PE apoptosis kit (BD Biosciences,
Franklin Lakes, NJ, USA) was used according to the manufacturer’s
instructions. Briefly, U373, U251, TR-U373 and TR-U251 cells were
treated with TMZ for 5 days prior to being harvested and washed
with cold PBS. The washed cells were resuspended in Annexin V
binding buffer followed by staining with Annexin V-PE together with
7-AAD. The stained cells were then analyzed by FACS
(Becton-Dickinson, Franklin Lakes, NJ, USA) and the results were
analyzed using Cell Quest software (Becton-Dickinson).
Western blot analysis
Cells were serum-starved for 24 h and then
pre-stimulated with 20 ng/ml IL-6 for 20 min immediately prior to
harvesting. Protein lysates were subjected to 10% SDS-PAGE and the
resolved proteins were transferred electrophoretically onto
nitrocellulose membranes (Pall, Pensacola, FL, USA). Membranes were
blocked with 5% skim milk and incubated with specific antibodies;
anti-O6-methylguanine-DNA-methyltransferase (MGMT;
Chemicon, Temecula, CA, USA), anti-STAT3 (Santa Cruz Biotechnology,
Santa Cruz, CA, USA), anti-phospho-STAT3 Y705 (Cell Signaling,
Beverly, MA, USA), anti-phospho-STAT3 S727 (Cell Signaling) and
anti-β-actin (Sigma-Aldrich). The blots were developed using a
chemiluminescence detection system (Amersham Biosciences,
Piscataway, NJ, USA).
STAT3 small interfering RNA
transfection
U373, U251, TR-U373 and TR-U251 cells were
transfected with STAT3 small interfering RNA (siRNA; 80 nM) or
control siRNA (80 nM) using Lipofectamine RNAiMAX (Invitrogen,
Carlsbad, CA, USA). Briefly, one day prior to transfection, the
cells (1×103) were either plated in 96-well plates to
determine cell viability or on 60 mm-diameter dishes
(1×105) for Western blot analysis with 10% FBS DMEM
(WelGene) without antibiotics. siRNA-lipofectamine complex mixture
in serum-free Opti-MEM (Gibco, Grand Island, NY, USA) was prepared
according to the manufacturer’s instructions. The medium was
replaced with DMEM containing 10% FBS 4 h after transfection. After
48 h, the protein extracts were prepared for Western blot analysis
and the cell viability was measured following additional treatment
with TMZ for 72 h.
Results
Generation of temozolomide-resistant
glioma cell lines
TR-U373 and TR-U251 cells were generated from the
parental cell lines by treatment with 2-fold increasing TMZ
concentrations (12.5–800 μM TMZ) every two passages over a period
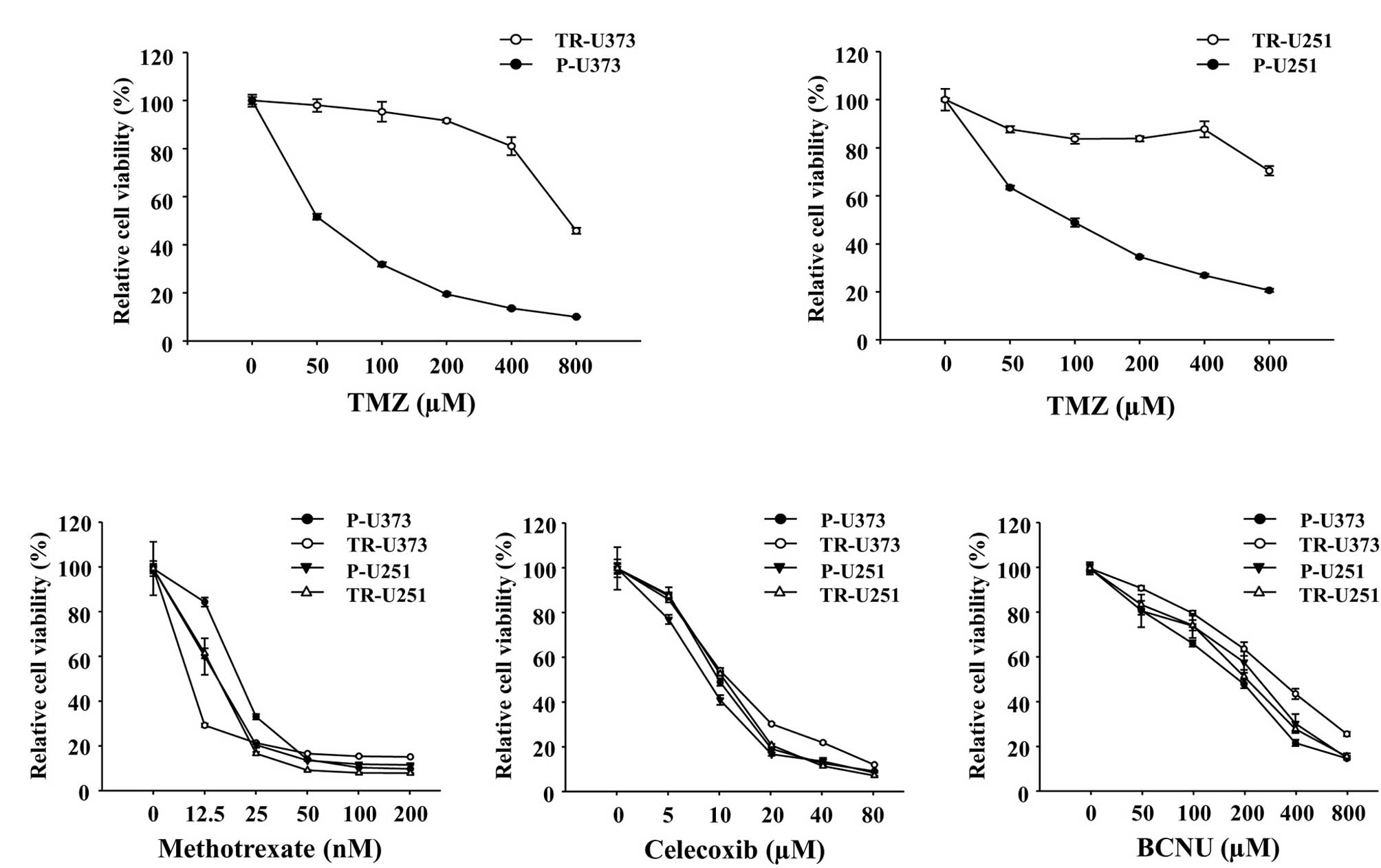
of 2 months. TMZ-resistance of the established TR cells was
evaluated by viability analysis using a CCK-8 assay. TR-U373 and
TR-U251 were 16- and 10-fold, respectively, more resistant to TMZ
as compared with the parental cells (IC50, 800 μM vs. 50
μM and 1,000 μM vs. 100 μM, respectively; Fig. 1A). TMZ-resistance of the TR cells
was maintained with the absence of drugs for almost 2 months (data
not shown). In addition, the TR cells exhibited no resistance to
various anti-cancer drugs including MTX, Celecoxib, and BCNU
(Fig. 1B).
Resistance to temozolomide-induced
apoptosis in TR-U373 and TR-U251 cells
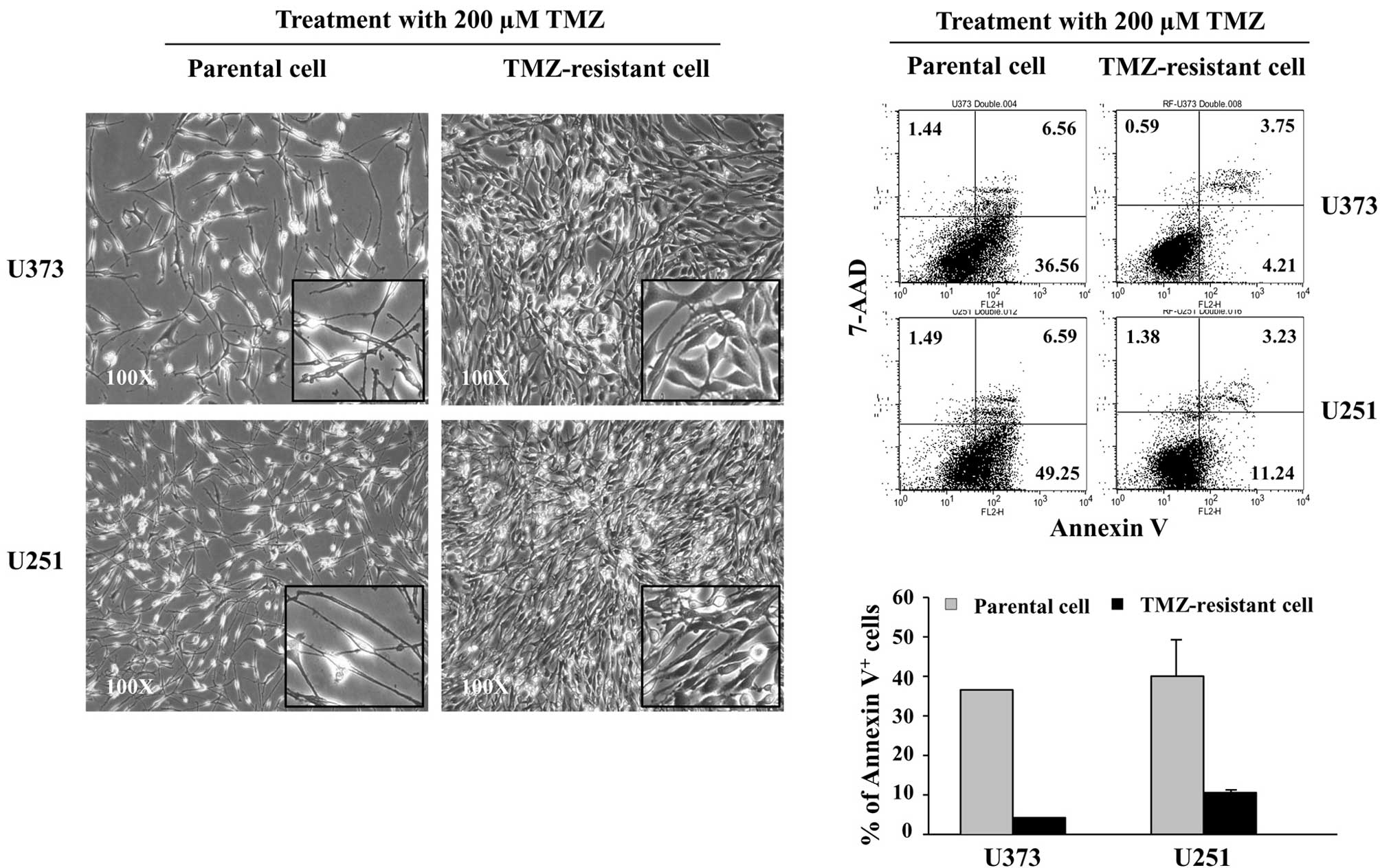
Following exposure to 200 μM TMZ for 5 days, the
cellular morphology of the TR cells was stably altered as compared
with the parental cells (Fig. 2A).
Since TMZ causes cell death through apoptosis (18), TMZ-induced apoptosis was compared
between parental and TR cells. Apoptosis induced by 200 μM TMZ was
evaluated with Annexin V/7-AAD staining by FACS. Following exposure
to TMZ, the early apoptotic Annexin V+/7-AAD−
cells were noted to have decreased in TR-U373 and TR-U251 cells
compared to those of the parental cells, from 37 to 4% and 49 to
11%, respectively. However, the incidence of necrotic Annexin
V−/7-AAD+ or late apoptotic Annexin
V+/7-AAD+ cells showed no significant
differences (Fig. 2B). The results
showed that TR cells are resistant to TMZ-induced apoptosis.
Temozolomide-resistance and MGMT
expression
The MGMT DNA repair protein contributes to drug
resistance through the demethylation of methyl adducts formed by
TMZ (19). To investigate whether
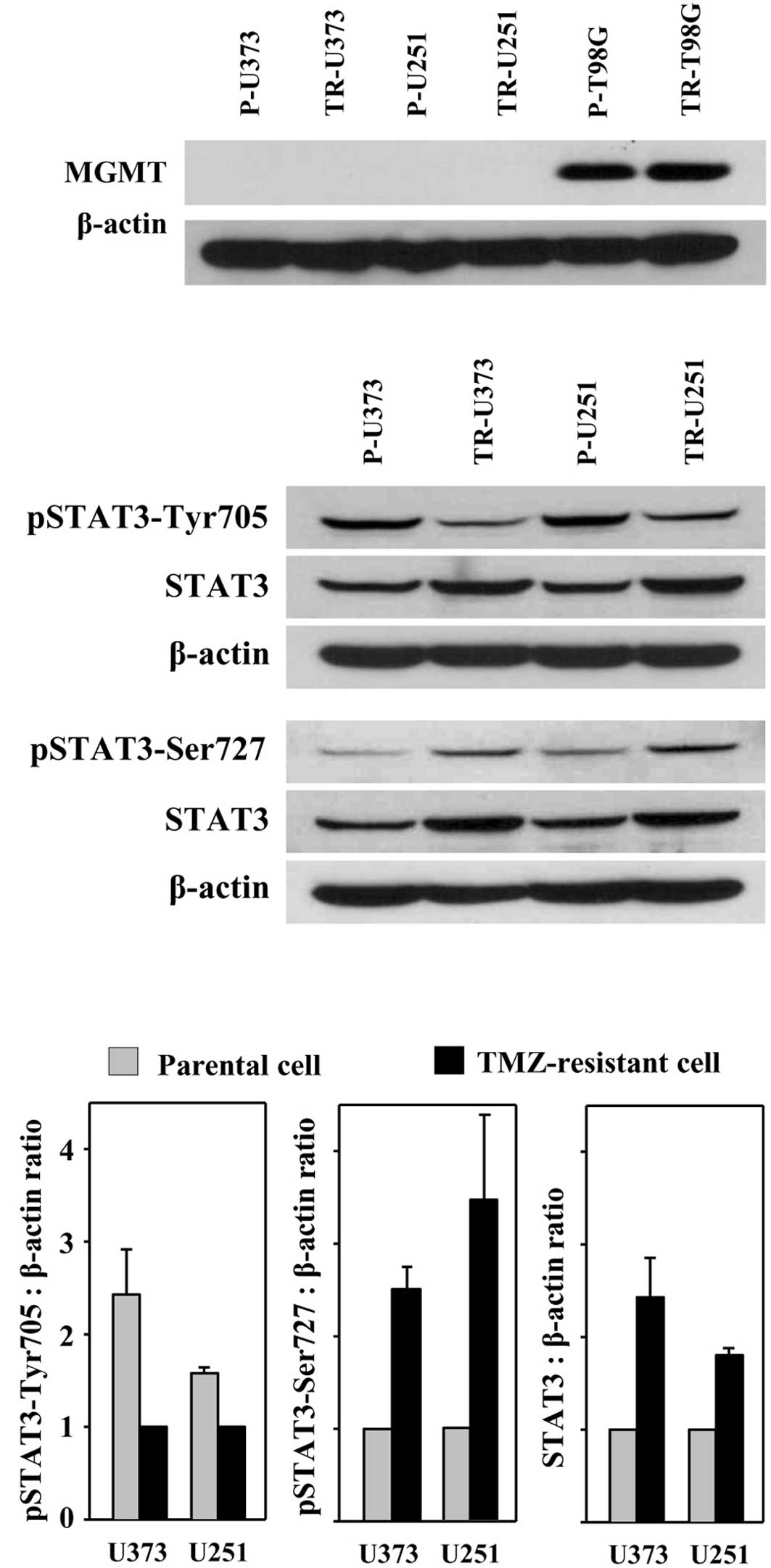
TMZ-resistance occurring in TR cells is due to the expression of
MGMT, the expression of MGMT in TR cells derived from MGMT
non-expressing U373 and U251 cells was assessed. Western blot
analysis detected MGMT expression in T98G and TR-T98G cells, but
not in U373, U251, TR-U373 and TR-U251 cells (Fig. 3A). Accordingly, TR cells showed a
strong resistance to TMZ, although MGMT expression was not
detected. The results suggest that TMZ resistance acquired in TR
cells is related to other molecules or factors.
Increased activation and expression of
STAT3 in temozolomide-resistant glioma cell lines
STAT3 is highly expressed and activated in
paclitaxel-resistant ovarian cancer cells (17). To determine whether the expression
and activation of STAT3 increased in TR cells, the expression level
of STAT3 and phosphorylated STAT3 were investigated in the parental
and TR cells by Western blot analysis. The levels of STAT3 and
pSTAT-Ser727 were increased in the TR cells as compared with the
parental cells. On the other hand, the level of pSTAT3-Tyr705 was
decreased (Fig. 3B). TR-U373 and
TR-U251 cells exhibited an ~2.4- and 1.8-fold increased level of
STAT3, respectively, and an ~2.3- and 2.9-fold increased level of
pSTAT3-Ser727, respectively, whereas the level of pSTAT3-Tyr705 was
decreased by ~2.4- and 1.6-fold, respectively (Fig. 3C).
Reversal of temozolomide-resistance using
small interfering RNA in TR-U251 cells
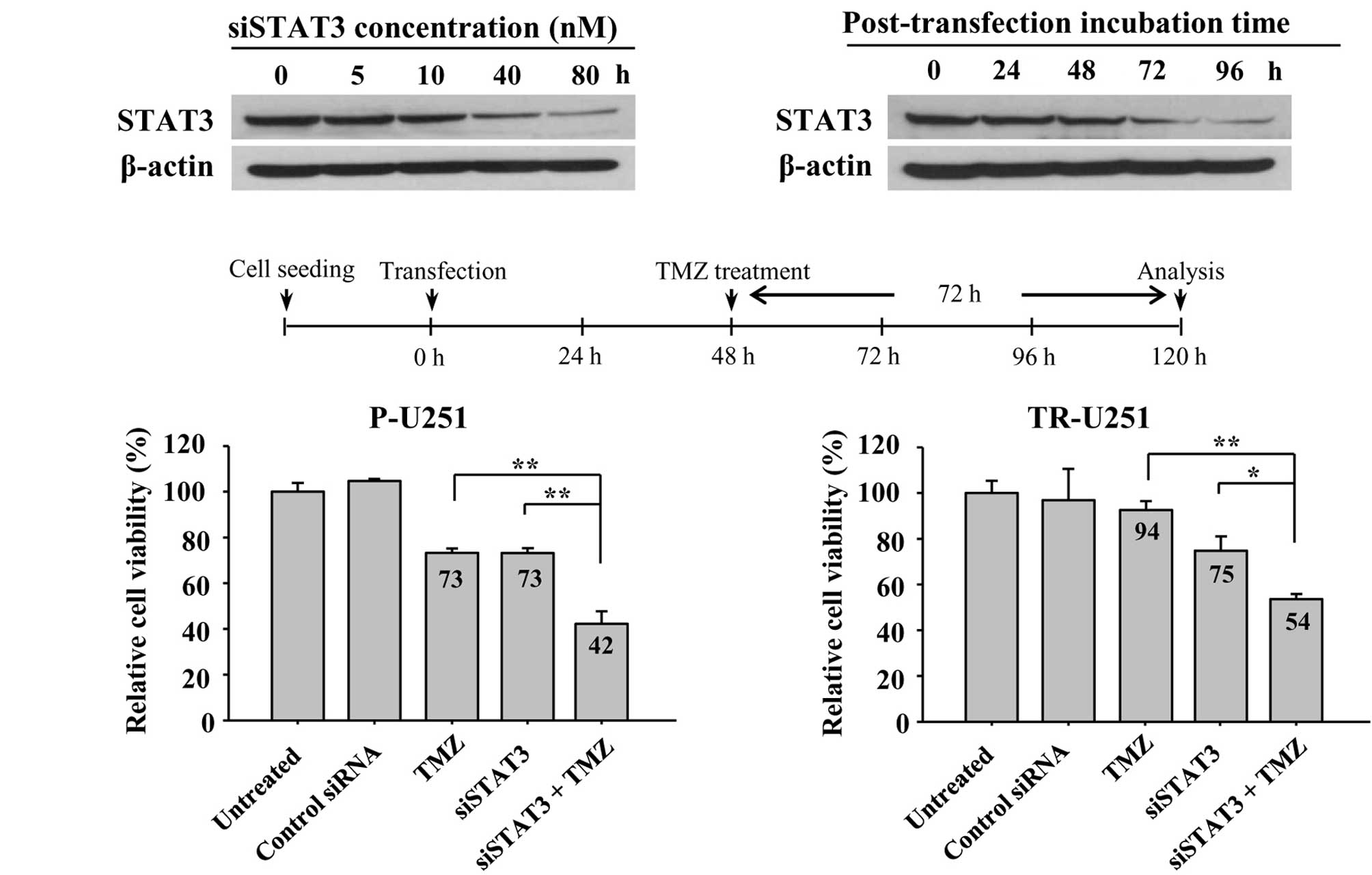
Based on the observation that the level of STAT3 is
elevated in TR cells, an experiment was performed to assess whether
the inhibition of STAT3 expression would increase TMZ sensitivity
in TR cells. First, to evaluate the knockdown efficiency of siRNA,
TR-U251 cells were treated with various concentrations of STAT3
siRNA. STAT3 expression was dose-dependently inhibited by STAT3
siRNA and its efficacy was maximized at 80 nM (Fig. 4A, left panel). In addition, to
determine the optimal incubation time after post-transfection based
on the experimental conditions, the expression of STAT3 was
determined at various time points following transfection by Western
blotting. Knockdown of STAT3 expression by siRNA was optimized at
96 h and maintained for 96 h (Fig.
4A). Based on these conditions, the effects of STAT3 knockdown
and TMZ treatment were determined. Both parental-U251 (P-U251) and
TR-U251 cells were transfected with STAT3 siRNA for 48 h and then
incubated in the absence or presence (200 μM) of TMZ for a further
72 h. Co-treatment with STAT3 siRNA and TMZ resulted in synergistic
decreases in cell viability as compared to each treatment alone
(Fig. 4B). Notably, TMZ-resistant
TR-U251 cells showed a relatively comparable effect on combination
treatment compared to TMZ-sensitive P-U251 cells. This result was
consistent with the association of STAT3 with TMZ-resistance,
thereby indicating that the inhibition of STAT3 is able to reverse
TMZ-resistance in TR cells.
Discussion
TMZ is considered to be the most effective drug in
the treatment of glioma. TMZ is also regarded as the standard
chemotherapeutic drug in combination with surgery and radiotherapy
(2). However, its efficacy is often
limited by tumor recurrence due to the development of resistance to
TMZ (3). Since the mechanism behind
TMZ-resistance development is unclear, the present study was
undertaken in order to generate TR cells to determine the molecular
mechanism of TMZ-resistance development.
TR cells were generated by exposure to a gradually
increasing TMZ concentration. Analysis of TMZ-resistance in TR
cells indicated that the resistant cells exhibit a strong
resistance to TMZ and have a significantly diminished progression
of apoptosis compared with the parental cell lines (Figs. 1 and 2). Such resistance was stable upon the
removal of TMZ and was maintained for a considerable period of
time. Earlier studies reported that cancer cells exposed to an
anticancer drug show resistance to diverse anticancer drugs that
are structurally and functionally different from the primary
anticancer drug (20,21). In contrast, the TR cells used showed
resistance to TMZ but not to other tested drugs, when compared with
the parental cells (Fig. 1). These
observations are in agreement with the previously reported result
that the viability of BCNU-resistant leukemia cells is subject to
inhibition by TMZ (22). Thus, it
can be considered that TR cells that develop resistance solely to
TMZ are a more useful model for the investigation of TMZ-resistance
in glioma cells.
TMZ is converted to monomethyl triazeno imidazole
carboxamide in the body, which in turn induces apoptosis in glioma
cells by methylating the O6-position of guanine.
However, this effect of TMZ is obstructed by MGMT which
demethylates the methyl adduct formed by TMZ (19). MGMT, which is expressed in a
subgroup of gliomas, plays a key role in cellular resistance to
alkylating agents including TMZ, BCNU and cyclophosphamide
(23). A real-time PCR analysis
revealed a strong expression of the gene encoding MGMT in
BCNU-resistant leukemia cells as compared with no detectable MGMT
gene expression in parental leukemia cells (22). However, in this study, MGMT was not
expressed in either parental or U373 and U251 TR cells. Moreover,
the expression of MGMT in T98G cells did not differ between the
parental and TR cells (Fig. 3A).
These observations provide evidence that other molecules or factors
affect the resistance to TMZ.
Previous studies reported that STAT3 is associated
with drug resistance as well as tumor cell proliferation and
apoptosis. STAT3-dependent overexpression of Bcl-2 confers a
survival advantage to breast cancer cells and contributes to their
chemoresistance (24). The
expression and activation of STAT3 is significantly increased in
paclitaxel-resistant ovarian cancer cells and tissues, and the
inhibition of STAT3 reduces paclitaxel resistance (17,25).
Similar findings are reported in DOX-resistant human cancer cells
including breast and liver cancer and neuroblastoma (26,27).
However, the role of STAT3 in the acquisition of TMZ-resistance in
glioma has yet to be determined. Therefore, the present study
investigated the relationship between STAT3 and TMZ-resistance in
glioma cells. STAT3 and pSTAT3-Ser727 were overexpressed in TR
cells as compared with the parental cells, whereas the expression
of pSTAT3-Tyr705 was decreased in TR cells (Fig. 3B).
With regard to STAT3 activity, it was reported that
as STAT3-Tyr705 is activated, drug resistance is increased
accordingly (17,27). STAT3 is activated by phosphorylation
on tyrosine residue 705 in response to cytokines such as IL-6 to
form a homodimer (28). This
activation transfers the expression of genes such as Bcl-2,
survivin and cyclin D1. STAT3 may be correlated to drug resistance
through tyrosine phosphorylation (24–26).
In the present study, however, TMZ-resistance was increased in a
viability assay while pSTAT-Tyr705 was decreased in TR cells
(Fig. 1A). When the cells were
treated with IL-6 to stimulate STAT3, Western blot analysis showed
a decreased expression of pSTAT3-Tyr705 in TR cells compared to the
parental cells. It is conceivable that the phosphorylation of STAT3
on tyrosine residue 705 in TR cells may not be stimulated by IL-6,
and pSTAT3-Tyr705 may not be directly associated with
TMZ-resistance acquired in TR cells.
STAT3 has another phosphorylation site on the serine
727 residue phosphorylated in response to IL-6 and other factors
(8,29). Serine phosphorylation is mediated
either directly or indirectly by a signal transduction cascade
involving MAPK, mTOR and PKCδ (11,30,31),
and it appears to regulate transactivation with tyrosine
phosphorylation. Recent studies have proposed that serine 727
phosphorylation is sufficient to activate STAT3 without tyrosine
705 phosphorylation, as is noted, for example, in the activation of
STAT3 in macrophages (32) or
chronic lymphocytic leukemia cells (33). Moreover, the activation of STAT3
through serine phosphorylation promotes prostate tumorigenesis
independent of tyrosine phosphorylation (34). These reports are in agreement with
our result in that the expression of pSTAT3-Ser727 was increased in
TR cells as compared with the parental cells. In addition,
phosphorylation of STAT3 on the serine 727 residue is enhanced
during mitosis and this correlates with a reduction of tyrosine 705
phosphorylation (35,36). Therefore, serine phosphorylation
negatively modulates tyrosine phosphorylation. Taken together, the
results suggest that STAT3 phosphorylation on the serine 727
residue is closely related with TMZ-resistance in TR cells.
STAT3 is transactivated by either phosphorylated or
unphosphorylated STAT3 (U-STAT3 or total STAT3) (37,38).
U-STAT3 accumulates in the nucleus by binding to unphosphorylated
NF-κB, and this complex, as a novel transcription factor, activates
(by a novel mechanism distinct from that used by STAT3 dimers) the
transcripton of genes including E2F-1, RANTES,
IL-6, IL-8 or MRAS. The products of
E2F-1-regulated genes, including cyclin D1, cyclin E,
cyclin B1 and cdc2, contribute to the cell cycle
transition and inhibit apoptosis. Notably, NF-κB is constitutively
activated in glioma cells (39);
consistent with this, the expression of U-STAT3 was found to be
increased in TR cells. Therefore, TMZ-resistance observed in the TR
glioma cells may be due to the overexpression of U-STAT3. This
suggestion agrees with the observation in this study that
siRNA-mediated inhibition of STAT3 reversed TMZ-resistance in TR
cells. Further molecular studies are therefore required to
elucidate the mechanisms regarding the relationship between
TMZ-resistance and pSTAT3-Tyr705, pSTAT3-Ser727 and U-STAT3.
In conclusion, STAT3 activation and expression was
found to be elevated in TR cells. Moreover, the inhibition of STAT3
expression results in an increased TMZ sensitivity of TR cells.
Thus, STAT3 is a potential molecular target for the development of
a novel strategy to inhibit TMZ-resistance in gliomas.
Acknowledgements
This study was supported by a grant from the
National R&D Program for Cancer Control, Ministry for Health,
Welfare and Family Affairs, Republic of Korea (0720330).
References
|
1
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yung WK, Prados MD, Yaya-Tur R, et al:
Multicenter phase II trial of temozolomide in patients with
anaplastic astrocytoma or anaplastic oligoastrocytoma at first
relapse. Temodal Brain Tumor Group. J Clin Oncol. 17:2762–2771.
1999.PubMed/NCBI
|
|
4
|
Balmaceda C, Peereboom D, Pannullo S, et
al: Multi-institutional phase II study of temozolomide administered
twice daily in the treatment of recurrent high-grade gliomas.
Cancer. 112:1139–1146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chamberlain MC and Johnston S: Salvage
chemotherapy with bevacizumab for recurrent alkylator-refractory
anaplastic astrocytoma. J Neurooncol. 91:359–367. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Butowski N, Lamborn KR, Lee BL, et al: A
North American brain tumor consortium phase II study of poly-ICLC
for adult patients with recurrent anaplastic gliomas. J Neurooncol.
91:183–189. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fabrini MG, Silvano G, Lolli I, et al: A
multi-institutional phase II study on second-line Fotemustine
chemotherapy in recurrent glioblastoma. J Neurooncol. 92:79–86.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wen Z, Zhong Z and Darnell JE Jr: Maximal
activation of transcription by Stat1 and Stat3 requires both
tyrosine and serine phosphorylation. Cell. 82:241–250. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu CL, Meyer DJ, Campbell GS, et al:
Enhanced DNA-binding activity of a Stat3-related protein in cells
transformed by the Src oncoprotein. Science. 269:81–83. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yokogami K, Wakisaka S, Avruch J and
Reeves SA: Serine phosphorylation and maximal activation of STAT3
during CNTF signaling is mediated by the rapamycin target mTOR.
Curr Biol. 10:47–50. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang YW, Wang LM, Jove R and Vande Woude
GF: Requirement of Stat3 signaling for HGF/SF-Met mediated
tumorigenesis. Oncogene. 21:217–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bromberg J: Stat proteins and oncogenesis.
J Clin Invest. 109:1139–1142. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Catlett-Falcone R, Landowski TH, Oshiro
MM, et al: Constitutive activation of Stat3 signaling confers
resistance to apoptosis in human U266 myeloma cells. Immunity.
10:105–115. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Duan Z, Foster R, Bell DA, et al: Signal
transducers and activators of transcription 3 pathway activation in
drug-resistant ovarian cancer. Clin Cancer Res. 12:5055–5063. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
D’Atri S, Tentori L, Lacal PM, et al:
Involvement of the mismatch repair system in temozolomide-induced
apoptosis. Mol Pharmacol. 54:334–341. 1998.PubMed/NCBI
|
|
19
|
Margison GP, Povey AC, Kaina B and
Santibanez Koref MF: Variability and regulation of
O6-alkylguanine-DNA alkyltransferase. Carcinogenesis. 24:625–635.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi CH: ABC transporters as multidrug
resistance mechanisms and the development of chemosensitizers for
their reversal. Cancer Cell Int. 5:302005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stavrovskaya AA: Cellular mechanisms of
multidrug resistance of tumor cells. Biochemistry (Mosc).
65:95–106. 2000.PubMed/NCBI
|
|
22
|
Yamauchi T, Ogawa M and Ueda T:
Carmustine-resistant cancer cells are sensitized to temozolomide as
a result of enhanced mismatch repair during the development of
carmustine resistance. Mol Pharmacol. 74:82–91. 2008. View Article : Google Scholar
|
|
23
|
Pegg AE, Dolan ME and Moschel RC:
Structure, function, and inhibition of O6-alkylguanine-DNA
alkyltransferase. Prog Nucleic Acid Res Mol Biol. 51:167–223. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Real PJ, Sierra A, De Juan A, Segovia JC,
Lopez-Vega JM and Fernandez-Luna JL: Resistance to chemotherapy via
Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer
cells. Oncogene. 21:7611–7618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Duan Z, Ames RY, Ryan M, Hornicek FJ,
Mankin H and Seiden MV: CDDO-Me, a synthetic triterpenoid, inhibits
expression of IL-6 and Stat3 phosphorylation in multi-drug
resistant ovarian cancer cells. Cancer Chemother Pharmacol.
63:681–689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim JH, Lee SC, Ro J, Kang HS, Kim HS and
Yoon S: Jnk signaling pathway-mediated regulation of Stat3
activation is linked to the development of doxorubicin resistance
in cancer cell lines. Biochem Pharmacol. 79:373–380. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rebbaa A, Chou PM and Mirkin BL: Factors
secreted by human neuroblastoma mediated doxorubicin resistance by
activating STAT3 and inhibiting apoptosis. Mol Med. 7:393–400.
2001.PubMed/NCBI
|
|
28
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: a
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wen Z and Darnell JE Jr: Mapping of Stat3
serine phosphorylation to a single residue (727) and evidence that
serine phosphorylation has no influence on DNA binding of Stat1 and
Stat3. Nucleic Acids Res. 25:2062–2067. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kovarik P, Stoiber D, Eyers PA, et al:
Stress-induced phosphorylation of STAT1 at Ser727 requires p38
mitogen-activated protein kinase whereas IFN-gamma uses a different
signaling pathway. Proc Natl Acad Sci USA. 96:13956–13961. 1999.
View Article : Google Scholar
|
|
31
|
Jain N, Zhang T, Kee WH, Li W and Cao X:
Protein kinase C delta associates with and phosphorylates Stat3 in
an interleukin-6-dependent manner. J Biol Chem. 274:24392–24400.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu H, Ma Y, Cole SM, et al: Serine
phosphorylation of STAT3 is essential for Mcl-1 expression and
macrophage survival. Blood. 102:344–352. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hazan-Halevy I, Harris D, Liu Z, et al:
STAT3 is constitutively phosphorylated on serine 727 residues,
binds DNA, and activates transcription in CLL cells. Blood.
115:2852–2863. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qin HR, Kim HJ, Kim JY, et al: Activation
of signal transducer and activator of transcription 3 through a
phosphomimetic serine 727 promotes prostate tumorigenesis
independent of tyrosine 705 phosphorylation. Cancer Res.
68:7736–7741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shi X, Zhang H, Paddon H, Lee G, Cao X and
Pelech S: Phosphorylation of STAT3 serine-727 by cyclin-dependent
kinase 1 is critical for nocodazole-induced mitotic arrest.
Biochemistry. 45:5857–5867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chung J, Uchida E, Grammer TC and Blenis
J: STAT3 serine phosphorylation by ERK-dependent and -independent
pathways negatively modulates its tyrosine phosphorylation. Mol
Cell Biol. 17:6508–6516. 1997.
|
|
37
|
Yang J, Liao X, Agarwal MK, Barnes L,
Auron PE and Stark GR: Unphosphorylated STAT3 accumulates in
response to IL-6 and activates transcription by binding to
NFkappaB. Genes Dev. 21:1396–1408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang J, Chatterjee-Kishore M, Staugaitis
SM, et al: Novel roles of unphosphorylated STAT3 in oncogenesis and
transcriptional regulation. Cancer Res. 65:939–947. 2005.PubMed/NCBI
|
|
39
|
Raychaudhuri B, Han Y, Lu T and Vogelbaum
MA: Aberrant constitutive activation of nuclear factor kappaB in
glioblastoma multiforme drives invasive phenotype. J Neurooncol.
85:39–47. 2007. View Article : Google Scholar : PubMed/NCBI
|