Introduction
Thyroid cancer represents approximately 2% of the
malignancies occurring in the US, accounting for an estimated
37,200 cancer diagnoses and 1,630 cancer-related deaths per year
(1). Of these cancers, 2–3% are
medullary thyroid cancer (MTC) (2,3). In
the literature, sporadic carcinomas are described in 48–86% of
patients and hereditary carcinomas in 14–52% of patients with MTC
(4). In the familial forms of MTC,
multicentric carcinomas were reported in 56–85% of patients. Both
sporadic and hereditary types of MTC metastasize to the cervical
lymph nodes in 68–80% of patients. The 5- and 10-year survival for
medullary carcinomas is 65–89% and 71–87%, respectively (5). Average survival for MTC is lower than
that for more common thyroid cancers, e.g., 83% 5-year survival for
MTC compared to 90–94% 5-year survival for papillary and follicular
thyroid cancer (6). Survival is
correlated with stage at diagnosis, and decreased survival in MTC
can be partly accounted for by a high proportion of late-stage
diagnoses (7). A Surveillance,
Epidemiology and End Results (SEER) population-based study of 1,252
MTC patients found that survival varied by the extent of local
disease. For example, the 10-year survival rates ranged from 95.6%
for disease confined to the thyroid gland to 40% for patients with
distant metastases (8).
MTC arises from parafollicular calcitonin-secreting
cells of the thyroid gland. MTC occurs in sporadic and familial
forms and may be preceded by C-cell hyperplasia (CCH), although CCH
is a relatively common abnormality in middle-aged adults. In a
population-based study in Sweden, 26% of patients with MTC
presented with the familial form (9). A French national registry and a US
clinical series both reported a higher proportion of familial cases
(43 and 44%, respectively) (10).
Familial cases often indicate the presence of multiple endocrine
neoplasia type 2, a group of autosomal dominant genetic disorders
caused by inherited mutations in the RET proto-oncogene.
MTC is a malignancy arising from the parafollicular
or C-cells of neuroendocrine origin. MTC was first described by
Jaquet in the German literature as ‘malignant goiter with amyloid’
(11). C-cells are named due to
their calcitonin hormone secretion and account for up to 1% of
thyroid cells. These cells are found throughout the thyroid gland
but are mostly located in the posterior upper third of the lateral
lobes, where the majority of MTCs are found. C-cells also produce
carcinoembryonic antigen (CEA) (12).
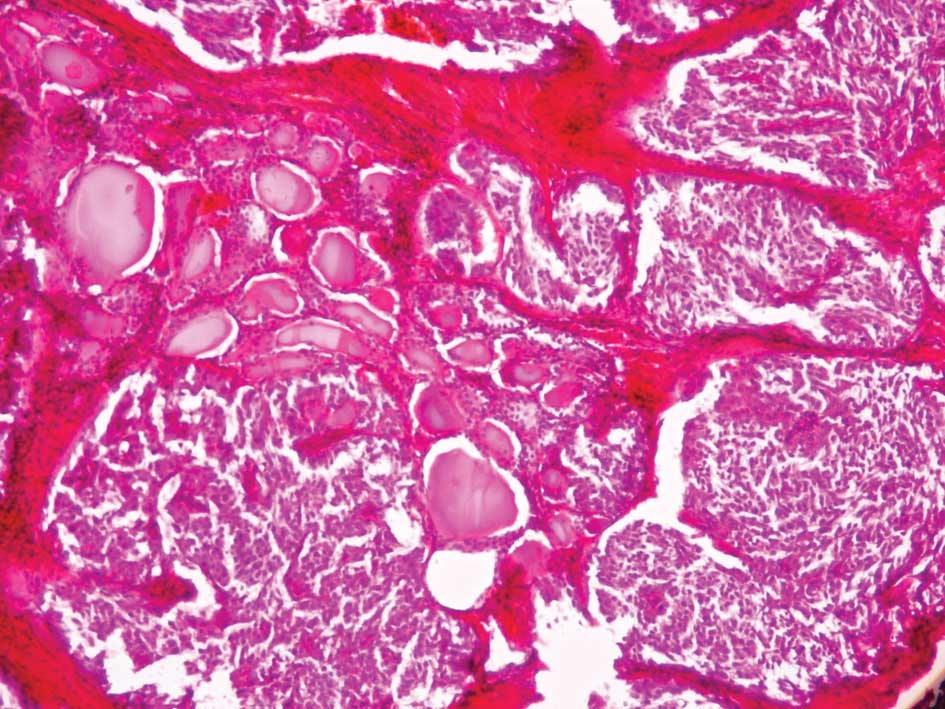
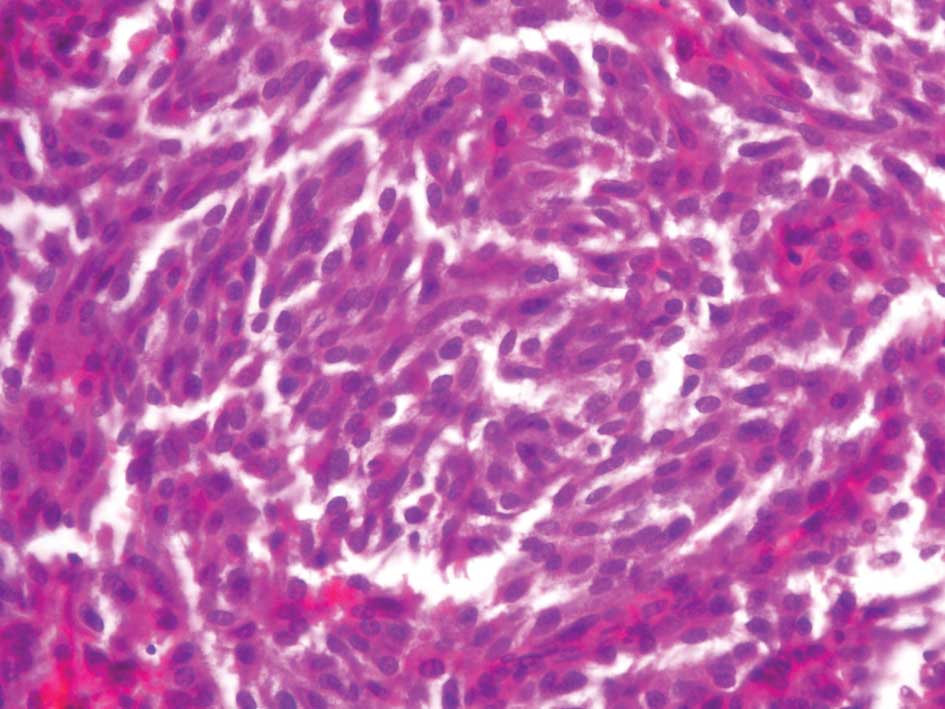
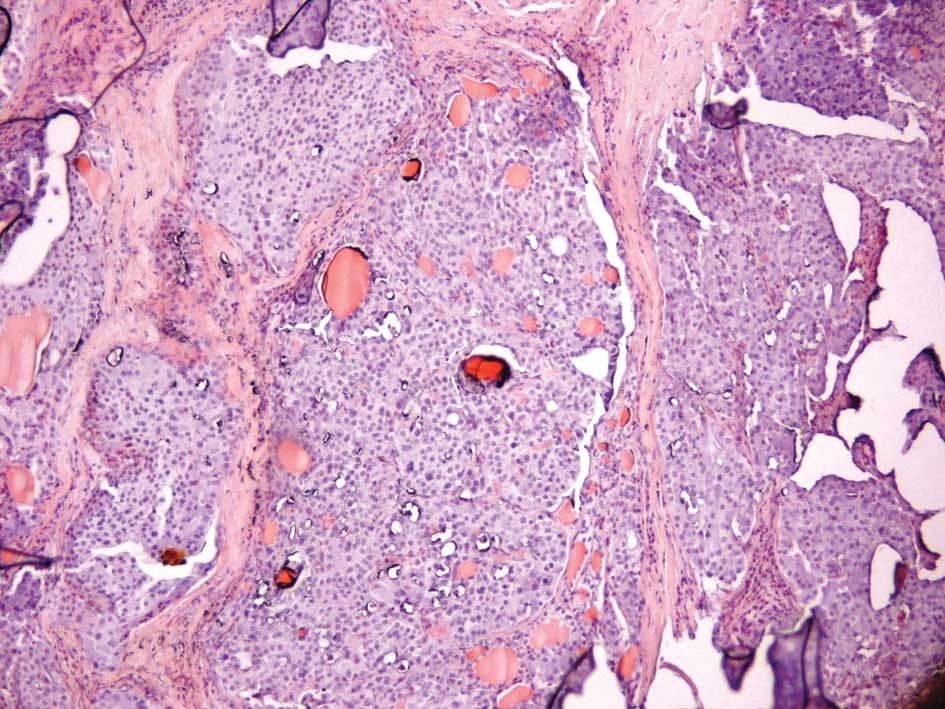
MTCs usually have a slow growth rate and appear gray
and firm. Histological reports commonly describe uniform polygonal
cells with central nuclei and finely granular eosinophilic
cytoplasm (Figs. 1 and 2). Stromal amyloid is found in one third
of MTCs (Fig. 3). Moreover, C-cell
hyperplasia is a precursor of malignant transformation (13). Approximately 75% of MTCs are
sporadic neoplasms (s-MTC), while 25% are familial, inherited
through an autosomal dominant pattern and associated with germline
mutations in the Ret proto-oncogene on chromosome 10q11.2.
This gene encodes a tyrosine kinase receptor protein (ret), with an
extracellular domain containing a ligand-binding site and a
cysteine-rich region, a transmembrane domain, and an intracellular
tyrosine kinase domain (14).
Familial MTC may occur as part of multiple endocrine neoplasia type
2A (MEN 2A) (MTC, pheochromocytoma and parathyroid hyperplasia) or
type 2B (MEN 2B) (MTC, pheochromocytoma, mucosal and alimentary
tract neuromas and marfanoid habitus) or without the presence of
any other endocrinopathies (familial non-MEN MTC) (15). Regarding the clinical presentation,
the most common symptom of MTC is a palpable thyroid nodule at
physical examination. Unfortunately, cervical lymph node metastasis
is commonly present by this time; up to 70%, with 10–15% of
patients exhibiting distant metastasis in the liver, lung, bone and
brain (16). The thyroid nodule is
associated with clinical symptoms, such as dysphagia, hoarseness,
dyspnea and coughing. Laryngoscopy usually reveals vocal cord
dysfunction resulting from involvement of the recurrent laryngeal
nerve. Patients with high calcitonin levels present with diarrhea
as a presenting sign of MTC. The severe diarrhea noted in some
patients with advanced extensive disease has been attributed to
prostaglandin secretion by the tumor. Other symptoms of ectopic
hormone production are facial flushing and, more rarely, Cushing’s
syndrome. Plasma calcitonin can be used for the diagnosis of s-MTC
and the preoperative diagnosis of C-cell hyperplasia (17). The most sensitive test for
preoperative diagnosis of MTC is fine needle aspiration (FNA)
biopsy, aided by immunocytochemical staining for calcitonin
(18).
Radiation treatment and conventional
chemotherapeutic regimens have been found to demonstrate no
improvement in the long-term survival for patients with MTC. Thus,
the optimal treatment is surgical management. The risk of
complications increases in reoperative procedures, thus the aim is
to perform a complete resection at the initial procedure. Since the
tumor is multifocal and bilateral in most patients with hereditary
MTC and in 20% of patients presenting with the sporadic type, the
treatment of choice and the safest option remains total
thyroidectomy (19).
Case report 1
A 56-year-old male patient was referred to our
Department for assessment. He was under endocrinological follow-up,
due to thyroid gland enlargement with a thyroid nodule in the right
lobe. The patient noted hoarseness of voice and neck enlargement.
He lacked other symptoms. Upon physical examination, the patient
appeared healthy with normal vital signs. No palpable lymph nodes
were noted, and the rest of the examination was normal. Laboratory
findings showed increased levels of plasma calcitonin, and an
ultrasound scan of the neck revealed enlargement of the thyroid
gland. A thyroid nodule was also found to be approximately 1 cm in
diameter in the right lobe, and nodes were absent. The patient
underwent FNA biopsy of the nodule where malignant cells were
found, indicating MTC. The patient underwent total thyroidectomy.
The thyroid gland was completely resected, and a thorough neck
examination did not reveal any nodes.
Cytopathologic examination showed a thyroid gland,
24 g total weight, which consisted of a 15-g right lobe with
dimensions 5.7×3.2×2 cm where multiple calcifications were
identified and a 9-mm diameter yellowish area was found; a 6-g left
lobe with dimensions of 3.8×2.3×1.7 cm where multiple
calcifications were also found; and the isthmus weighing 3 g with
dimensions 2.5×2.3×1 cm. Histologically, unilateral development of
the medullary thyroid cancer of 9 mm was found in the right lobe of
the conventional type with minor infiltration of the thyroid
sheath, grade pT1. The rest of the thyroid showed lesions of
thyroiditis. Postoperative radioactive scanning did not reveal any
residual thyroid. The patient is under medical treatment with
thyroxine per os for thyroid function substitution. The levels of
plasma calcitonin are within the normal range, and no recurrence
has been found on regular neck examinations during the two-year
follow-up following the initial operation.
Case report 2
A 48-year-old female patient was referred to our
Department for assessment. She was under endocrinological follow-up
due to autoimmune thyroiditis Hashimoto and was being treated with
thyroxine 0.1 mg per day per os. In the last two years, an increase
in the plasma calcitonin levels was noted, and the patient was
referred for surgical treatment. The patient exhibited no symptoms,
such as diarrhea, dysphagia or facial flashing. Physical
examination was normal. No palpable lymph nodes were detected, and
the rest of the examination was normal. A laboratory examination
showed increased levels of plasma calcitonin: 13.3 pg/ml (normal
values 0–10 pg/ml). Neck ultrasound scan revealed an increase in
the dimensions of the thyroid, a thyroid nodule ~1.5 cm in diameter
in the right lobe, five small nodules ~0.5 cm in diameter in the
left lobe and the absence of any nodes. The patient underwent total
thyroidectomy. The thyroid gland was resected completely, and a
thorough neck examination did not reveal any nodes.
Cytopathology revealed a thyroid gland, with a total
weight of 21.5 g, consisting of a right lobe of 3×4×2.5 cm where a
white nodule of 1.5 cm was found; and a left lobe with dimensions
4×2.5×1 cm where 3 calcificated areas were found equal to 0.1–0.4
cm and 2 colloidal nodules 0.4–0.6 cm were noted. Histologically,
the development of MTC was found in the left lobe in 3 sites with a
maximum diameter of 0.4 cm. The lymph nodes found were negative,
and the rest of the thyroid gland showed thyroiditis lesions. The
patient was treated with thyroxine per os for thyroid function
substitution. The plasma calcitonin levels were within the normal
range, and no recurrence was found upon a regular neck examination
at follow-up, four months after the initial operation.
Case report 3
A 49 year-old female patient was referred to our
Department for assessment. She underwent a thyroid panel scan due
to weight gain. No previous history of thyroid lesions had been
noted. The thyroid scan revealed normal thyroid hormonal values
along with increased calcitonin levels and traceable CEA antigen
levels. Moreover, a pentagastrin stimulation test was positive,
which in combination with the above-mentioned results, resulted in
a diagnosis of MTC. Total thyroidectomy was performed.
Cytopathology revealed a thyroid gland dispersed with multiple
nodules, along with notable nodes of 1 cm in diameter.
Histologically, the specimen was grade pT1. The plasma calcitonin
levels were within the normal range and no recurrence was found
upon a regular neck examination at follow-up, two months after the
initial operation.
Discussion
Medullary thyroid cancer is a rare aggressive type
of thyroid neoplasia. Significant predictors for MTC are age,
gender, clinical presentation, TNM stage, distant metastases and
extent of thyroidectomy. Primary and independent prognostic factors
are age and disease stage at the time of diagnosis. The recent Mt.
Sinai study showed 5-, 10- and 20-year overall survival rates as
97, 88 and 84%, respectively. Disease-free survival rates were 97,
74 and 29% at 5, 10 and 20 years, respectively (20).
MTC is a heterogeneous disease in terms of
biological behavior with a variable and unpredictable behavior. The
clinical course of MTC varies from an extremely indolent tumor that
can go unchanged for years to an aggressive variant that is
associated with a high mortality rate. MTC occurs either as a
sporadic event or secondary to a germline mutation with an
autosomal dominant pattern of inheritance. Unlike most other solid
tumors, the presence of microscopic residual disease within the
thyroid, local regional lymph nodes, or distant organs can be
detected by elevated serum levels of calcitonin or CEA. MTC
frequently metastasizes to regional lymph nodes, usually apparent
at the time of diagnosis. The frequency of nodal metastasis has
been reported to be more than 50% in patients with palpable
established primary tumors. The spread of metastases is most common
to the central compartment (level VI), followed by the ipsilateral
jugular chain of nodes (levels II–V) and the contralateral cervical
nodes. It may also be noted in the upper and anterior mediastinum.
Hematogenous spread to the lungs, liver, bones, brain and soft
tissues may occur. The fine military pattern of these metastases
renders conventional imaging a challenge. Laparoscopy may be useful
in the identification of small metastatic deposits in the liver of
patients with elevated calcitonin levels (21). Patients with MTC, without any lymph
node metastases, treated in the early stages of the disease, thus
have a low risk of recurrence. In contrast, patients with nodal
disease at presentation have a high risk for developing recurrent
or persistent disease. Follow-up is advised to commence 2–3 months
postoperatively with baseline calcitonin and CEA levels, and then
annually. Ultrasound of the neck had no proven benefit.
Thyroid-hormone replacement is required. Patients with hereditary
MTC are at risk of developing pheochromocytoma and
hyperparathyroidism (22,27).
The surgical treatment of MTC is determined by a
number of factors. The clinical course of MTC is usually more
aggressive than that of non-medullary differentiated thyroid
cancer, with high rates of recurrence and mortality, particularly
in young patients. Moreover, nodal metastases are present in more
than 70% of patients with palpable disease. Radiation treatment and
conventional chemotherapeutic regimens have exhibited no
improvement in long-term survival for patients with MTC. Thus, the
optimal treatment is surgical management. The risk of complications
increases in cases of recurrence, which require surgical
management, and the aim is to perform a complete resection at the
initial procedure. Since the tumor is multifocal and bilateral in
the majority of the patients with hereditary MTC and in 20% of
patients with the sporadic type, the treatment of choice and the
safest option remains total thyroidectomy. Preoperative measurement
of urine catecholamines is mandatory in order to exclude
pheochromocytoma (23). When
excessive catecholamine secretion is detected, adrenal surgery
should be performed in advance (24). In the literature, total
thyroidectomy and paratracheal dissection is proposed for patients
with MTC.
Lymph node metastases in patients with MTC may be
the first clinical sign. Approximately 20% of patients present with
distant metastases on diagnosis (25). As tumor stage increases, a higher
incidence of metastasis occurs in the contralateral central neck,
ipsilateral lateral neck and contralateral lateral neck
compartments. It is extremely uncommon for metastatic spread to
occur to the lateral nodes without first involving the ipsilateral
central nodes. Surgical excision is the only effective therapy for
these metastases (26).
Surgery has been the only generally effective
therapy for MTC. Unlike papillary thyroid cancer, there are only
limited options for patients with disseminated disease, and no
accepted adjuvant therapies are available. Radioactive iodine is
part of the standard treatment for papillary thyroid cancer, but
since C-cells are not of thyroid follicular origin, radioactive
iodine is not absorbed by C-cells. External beam radiation therapy
causes extensive scarring and fibrosis within the neck, limiting
surgical interventions. However, radiation therapy can be applied
for palliative reasons both for local disease and metastases to the
bones. Patients with metastatic disease can suffer severe symptoms
caused by calcitonin excess and may benefit from medical treatment
with somatostatin analogues. These patients may also benefit from
cytoreductive surgery of unresectable disease.
Conventional chemotherapy has been shown to have
limited efficacy in patients with MTC. Complete responses are rare,
and partial responses have been noted in less than one third of
patients. The side effect profile of chemotherapy is often adverse,
making this an unappealing option for many patients. Single-agent
regimens using doxorubicin, dacarbazine, capecitabine and
5-fluorouracil have been reported with partial response rates of up
to 24–29%. More novel chemotherapeutic agents, such as irinotecan,
a topoisomerase I inhibitor and 17-AAG, a heat-shock protein 90
(Hsp90) inhibitor, are currently being evaluated in phase II
clinical trials (28). Various
approaches aimed at developing systemic molecular therapies,
requiring targets specifically expressed by MTC cells, are
currently in progress (29). The
challenge for investigators is in analyzing the degree to which
Ret is being effectively inhibited and correlating the
results with surrogate markers and outcome data. Imitanib mesylate
(Gleevec®; Novartis Pharmaceuticals Corp., East Hanover,
NJ, USA) is a known tyrosine kinase inhibitor already in clinical
use against chronic myelogenous leukemia and gastrointestinal
stromal tumors, targeting specific tyrosine kinases. Studies
involving this drug performed in vitro have shown
dose-dependent inhibition of MTC cells and inhibition of
phosphorylation of the RET protein. Hsp90 is another
chemotherapeutic target that has been tested in MTC. In its normal
role, Hsp90 acts as one of a number of molecular chaperones,
facilitating normal cell proliferation and activity by binding to
specific signal-transduction proteins including the RET tyrosine
kinase receptor. Tumor cells overexpress active Hsp90, leading to
unregulated cell activity and proliferation. Drugs targeting
components of new pathways for Ret-negative tumors are currently
under trial, including angiogenesis inhibitors, proteasome
inhibitors, and cytotoxic chemotherapy in combination with tyrosine
kinase inhibitors or angiogenesis inhibitors. Potential therapeutic
targets include the manipulation of various cellular signaling
pathways, such as the PI3K-Akt, MAPK and Notch-1- hairy enhancer of
split (HES)-1-achaete-scute complex-like (ASCL)-1 signaling
pathway, and the glycogen synthase kinase-3 (GSK-3) pathway
(30).
In conclusion, MTC accounts for 5–10% of all thyroid
cancers. The majority of cases are sporadic, but 20% of cases are a
result of a germline mutation in the Ret proto-oncogene. The
management of medullary thyroid cancer is predominantly surgical
excision, consisting of a total thyroidectomy and lymph node
dissection. The extent and timing of surgical excision are crucial.
Systemic therapeutic options are limited for MTC. However, various
therapeutic targets show promise for the future development of new
therapies.
References
|
1
|
American Cancer Society. Cancer Facts and
Figures. Atlanta, Ga: American Cancer Society; 2009
|
|
2
|
National Cancer Institute. SEER Incidence:
Thyroid Cancer. Bethesda, MD: 2004
|
|
3
|
Hundahl SA, Fleming ID, Fremgen AM and
Menck HR: A National Cancer Data Base report on 53,856 cases of
thyroid carcinoma treated in the U.S., 1985–1995. Cancer.
83:2638–2648. 1998.PubMed/NCBI
|
|
4
|
Engelbach M, Gorges R, Forst T, et al:
Improved diagnostic methods in the follow-up of medullary thyroid
carcinoma by highly specific calcitonin measurements. J Clin
Endocrinol Metab. 85:18902000.
|
|
5
|
Safioleas M, Stamatakos M, Karampali E,
Rompoti N, Mouzopoulos G and Lygidakis N: Diagnosis and treatment
aspects of medullary thyroid carcinoma. Chirurgia. 101:121–126.
2006.
|
|
6
|
Bhattacharyya N: A population-based
analysis of survival factors in differentiated and medullary
thyroid carcinoma. Otolaryngol Head Neck Surg. 128:115–123. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Modigliani E, Vasen HM, Raue K, et al:
Pheochromocytoma in multiple endocrine neoplasia type 2: European
study. The Euromen Study Group. J Intern Med. 238:363–367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roman S, Lin R and Sosa JA: Prognosis of
medullary thyroid carcinoma: demographic, clinical, and pathologic
predictors of survival in 1252 cases. Cancer. 107:2134–2142. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bergholm U, Bergström R and Ekbom A:
Long-term follow-up of patients with medullary carcinoma of the
thyroid. Cancer. 79:132–138. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kebebew E, Ituarte PH, Siperstein AE, Duh
QY and Clark OH: Medullary thyroid carcinoma: clinical
characteristics, treatment, prognostic factors and a comparison of
staging systems. Cancer. 88:1139–1148. 2000. View Article : Google Scholar
|
|
11
|
Jaquet AJ: Ein fall von metastasierenden
amyloidtumoren (lymphosarcoma). Virchows Arch. 185:251–267. 1906.
View Article : Google Scholar
|
|
12
|
Rufini V, Castaldi P, Treglia G, et al:
Nuclear medicine procedures in the diagnosis and therapy of
medullary thyroid carcinoma. Biomed Pharmacother. 62:139–146. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fialkowski EA and Moley JF: Current
approaches to medullary thyroid carcinoma, sporadic and familial. J
Surg Oncol. 94:737–747. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeLellis RA: Pathology and genetics of
thyroid carcinoma. J Surg Oncol. 94:662–669. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matoso A, Zhou Z, Hayama R,
Flesken-Nikitin A and Nikitin AY: Cell lineage-specific
interactions between Men1 and Rb in neuroendocrine neoplasia.
Carcinogenesis. 29:620–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boikos SA and Stratakis CA: Molecular
mechanisms of medullary thyroid carcinoma: current approaches in
diagnosis and treatment. Histol Histopathol. 23:109–116.
2008.PubMed/NCBI
|
|
17
|
Evans DB, Burgess MA, Goepfert H and Gagel
RF: Medullary thyroid cancer. Current therapy in endocrinology and
metabolism. 6th edition. Bardin CW: Mosby-Year Book; St. Louis: pp.
127–132. 1997
|
|
18
|
Liu FH, Hsueh C, Chang HY, Liou MJ, Huang
BY and Lin JD: Sonography and fine-needle aspiration biopsy in the
diagnosis of benign versus malignant nodules in patients with
autoimmune thyroiditis. J Clin Ultrasound. 37:487–492. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sippel RS, Kunnimalaiyaan M and Chen H:
Current management of medullary thyroid cancer. Oncologist.
13:539–547. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Clark JR, Fridman TR, Odell MJ, Brierley
J, Walfish PG and Freeman JL: Prognostic variables and calcitonin
in medullary thyroid cancer. Laryngoscope. 115:1445–1450. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Toledo SP, Lourenço DM Jr, Santos MA,
Tavares MR, Toledo RA and Correia-Deur JE: Hypercalcitoninemia is
not pathognomonic of medullary thyroid carcinoma. Clinics.
64:699–706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lundgren CI, Delbridg L, Learoyd D and
Robinson B: Surgical approach to medullary thyroid cancer. Arq Bras
Endocrinol Metabol. 51:818–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oskam IM, Hoebers F, Balm AJ, et al: Neck
management in medullary thyroid carcinoma. Eur J Surg Oncol.
34:71–76. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dralle H and Machens A: Surgical
approaches in thyroid cancer and lymph-node metastases. Best Pract
Res Clin Endocrinol Metab. 22:971–987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Machens A, Schneyer U, Holzhausen HJ and
Dralle H: Prospects of remission in medullary thyroid carcinoma
according to basal calcitonin level. J Clin Endocr Metab.
90:2029–2034. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Al-Rawi M and Wheeler MH: Medullary
thyroid carcinoma – update and present management controversies.
Ann R Coll Surg Engl. 88:433–438. 2006.
|
|
27
|
Ball DW: Medullary thyroid cancer:
therapeutic targets and molecular markers. Curr Opin Oncol.
19:18–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
You YN, Lakhani V, Wells SA Jr and Moley
JF: Medullary thyroid cancer. Surg Oncol Clin N Am. 15:639–660.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schlumberger M, Carlomagno F, Baudin E,
Bidart JM and Santoro M: New therapeutic approaches to treat
medullary thyroid carcinoma. Nat Clin Pract Endocrinol Metab.
4:22–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cohen MS, Hussain HB and Moley JF:
Inhibition of medullary thyroid carcinoma cell proliferation and
RET phosphorylation by tyrosine kinase inhibitors. Surgery.
132:960–966. 2002. View Article : Google Scholar : PubMed/NCBI
|