Introduction
Cyclin D1 is a significant regulator of cell cycle
progression in numerous cell types. Cyclin D1 elicits its
pro-proliferative function early in the G1 phase, as it is capable
of activating cyclin-dependent kinases (CDKs) 4 or 6. Active
CDK4/6- cyclin D1 complexes phosphorylate and inactivate the
retinoblastoma protein (Rb), which is critical for modulating G1-
to S-phase progression, and in this manner promote cell
proliferation (1–3). In addition to its well-established
cell cycle roles, cyclin D1 is involved in CDK-independent function
in transcription by acting as a molecular bridge between DNA-bound
transcription factors and chromatin-modifying enzymes (4–6).
Cyclin D1 expression is regulated mainly by
extracellular mitogenic and oncogenic signals, allowing cyclin D1
to serve as a mediator of growth factor signaling and cell cycle
progression (7). It is therefore
unsurprising that cyclin D1 is often deregulated in tumors of
various origins (8). The
overexpression of cyclin D1 caused by gene amplification is
observed in several carcinomas, including those of the esophagus,
head and neck, breast and colon (9–16).
Notably, unlike strong oncogenes, such as Ras, the overexpression
of cyclin D1 alone is not capable of transforming immortalized
murine fibroblasts in vitro (17). Furthermore, whereas the
overexpression of cyclin D1 is considered to be the initial genetic
trigger in mantle cell lymphoma, targeted expression of wild-type
cyclin D1 in lymphoid cells does not result in a tumor-prone
phenotype in transgenic mice (18,19),
thereby challenging the notion that cyclin D1 is an oncogene.
Findings of a previous study showed that the
inhibition of cyclin D1 nuclear export during the S phase unmasks
its neoplastic potential (20).
Phosphorylation of cyclin D1 at threonine 286 (Thr286) is required
for its nuclear export and degradation in the cytoplasm (17,21,22).
This phosphorylation is mediated by glycogen synthase kinase 3β
(GSK-3β) and is greatly enhanced by the binding of cyclin D1 to
CDK4 (23). The expression of the
artificially engineered cyclins D1-T286A or D1b, a naturally
occurring alternative splice variant of cyclin D1, cannot be
phosphorylated by GSK-3β and are stabilized in the nucleus.
Moreover, these variants are capable of transforming murine
fibroblasts in the absence of a collaborating oncogene (21,24).
Furthermore, constitutively nuclear cyclin D1 mutants have been
identified in certain solid tissue tumors, such as esophageal and
endometrial cancers, that promote tumorigenesis in transgenic mice
(25,26). These results suggested that the
deregulation of cyclin D1 nuclear export is a tumor-initiating
event. Although cancer-derived cyclin D1 mutants are potent
oncogenes in vitro and in vivo, the mechanisms by
which they contribute to neoplasia remain to be elucidated. Among
the mutations detected in esophageal cancers was a deletion
encompassing codons 266–295 of cyclin D1 (Δ266–295). As with cyclin
D1b, this cancer-derived D1-Δ266–295 mutant possesses the cyclin
box required for CDK binding and enzymatic activity, but lacks the
PEST destruction box and Thr286, which are crucial to the promotion
of the nuclear export of cyclin D1 and its turnover (14). Using function analysis, the purpose
of this study was to show that this cancer-derived deletion mutant
D1-Δ266–295 retained its ability to support CDK4 catalytic
activity, and was characterized by constitutive nuclear
localization, thereby contributing to increasing cyclin D1
oncogenic capacity. Consequently, insight would be gained as to the
mechanism involved in such mutations contributing to the genesis
and progression of neoplastic growth.
Materials and methods
Cell culture conditions and
transfections
NIH3T3 and HepG2 cells were cultured in DMEM
supplemented with 2 mM L-glutamine, 10% FBS and antibiotics (Gibco,
Carlsbad, CA, USA). KYSE510 cells were grown with RPMI-1640
supplemented with 2 mM L-glutamine, 10% FBS and antibiotics.
Transient transfections were performed following the manufacturer’s
instructions using Lipofectamine Plus (Invitrogen, Carlsbad, CA,
USA). Cyclin D1-Δ266–295 plasmid was engineered using pFlex-cyclin
D1a vector as a template with primers
5′-GGTGGTGATTACAAAGATGACGACGATAAG-3′ (forward) and
5′-ACGGAATTCAGTTCTGCTGGGCCTG-3′ (reverse). The PCR products were
purified with Wizard® SV Gel and the PCR clean-up system
(Promega, Madison, WI, USA) and inserted into the pFlex vector as
EcoRI fragments as previously described for cyclin D1a
(24) to generate Flag-tagged
molecules.
Immunoblotting and immunoprecipitation
assays
For direct Western blot analysis, cells were washed
with pre-chilled phosphate-buffered saline (PBS) and lysed in lysis
buffer containing 0.1 mol/l NaCl, 0.01 mol/l Tris-Cl (pH 7.6), 0.1%
SDS, 20 mmol/l EDTA (pH 8.0), 10% Triton X-100, 1 mmol/l PMSF, 10
μg/ml aprotinin and 10 μg/ml leupeptin. Total cell protein was
resolved on denaturing polyacrylamide gels, transferred to
nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), and blotted
with antibodies obtained for cyclin D1a (SC-8396; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), M2 (F1804; Sigma, St. Louis,
MO, USA), Rb (9309L; CST), phospho-Rb-Ser780 (9307S; CST), Myc
(BM2901-02; Biomiga), Tublin (sc-9104; Santa Cruz Biotechnology)
and CDK4 (sc-26; Santa Cruz Biotechnology). Protein-antibody
complexes were visualized either by using secondary antibodies
(goat-anti-rabbit IgG or goat-anti-mouse IgG) followed by enhanced
chemiluminescence, or by using secondary antibodies conjugated with
Cy5.5 (Amersham Pharmacia Biotech, Piscataway, NJ, USA) and
visualized using the LI-COR Odyssey IR Imaging System (LI-COR
Biosciences, Lincoln, NE, USA).
For co-immunoprecipitation (Co-IP), cells were lysed
in IP buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM
EDTA, 1% Triton X-100, 10% glycerol, 1 mmol/l PMSF, 10 μg/ml
aprotinin and 10 μg/ml leupeptin, and were centrifuged for 30 min
at 10,000 × g at 4°C. The supernatant was incubated with primary
antibody overnight at 4°C. The immunocomplexes were bound to
protein-G sepharose 4B (Pharmacia, USA) for 1 h at 4°C and washed
three times with IP buffer. Proteins bound to the protein-G
sepharose 4B were eluted by adding Laemli-SDS sample buffer and
then boiling for 5 min. Following centrifugation at 10,000 × g for
2 min, the supernatant was analyzed by immunoblotting.
Protein turnover analysis
To measure the turnover rate of cyclin D1 protein,
Flag-tagged wild-type or mutant cyclin D1 plasmids were transiently
transfected into NIH-3T3 cell lines. Forty-eight hours after
transfection, cycloheximide (50 μg/ml; Sigma) was added to block
new protein synthesis. Cells were then harvested at 0, 30, 60, 90,
120 and 180-min intervals following treatment with cycloheximide.
Cell lysates were resolved by SDS-PAGE and the rate of cyclin D1
decay was then assayed by direct Western blotting.
Indirect immunofluorescence assays
NIH-3T3 cells overexpressing either Flag-tagged
wild-type or mutant cyclin D1 were seeded on glass coverslips.
Cells were then fixed using either 3% paraformaldehyde or
methanol-acetone (1:1) as previously described (24). Visualization of cyclin D1 was
achieved with the Flag-specific M2 monoclonal antibody. Secondary
TRITC-conjugated anti-mouse antibody (IgG; Sigma) staining was
performed for 60 min at room temperature under moisture. DNA was
visualized using Hoechst 33258 dye at a 1:1,000 dilution.
Coverslips were mounted on glass slides with vectashield medium
(Vector Laboratories Inc., Burlingame, CA, USA) and visualized
under a fluorescence microscope.
Real-time quantitative PCR analysis of
gene expression
RNA isolation was performed using standard
protocols. cDNA was prepared by reverse transcription (SuperScript,
Invitrogen). Real-time PCR was performed on a Roche
LightCycler® 480 sequence detection system using
LightCycler 480 SYBR-Green I Master mix (Roche, Indianapolis, IN,
USA). Amplification of the housekeeping gene GAPDH was performed to
standardize the amount of sample RNA. Primers used for the
detection of human Notch1 were 5′-CACTGATCCTGGCTGCC CGC-3′
(forward); and 5′-CAGCAGCACCTTGGCGGTCT-3′ (reverse). Primers used
for the detection of human GAPDH were 5′-GAGTCAACGGATTTGGTAGT-3′
(forward); and 5′-TTG ATTTTGGAGGGATCTCG-3′ (reverse). The thermal
cycler conditions were as follows: 2 min at 50°C, hold for 10 min
at 95°C, followed by two-step PCR for 40 cycles of 95°C for 15 sec
followed by 60°C for 1 min. After normalization to the GAPDH, the
relative quantification of gene expression was performed using the
2−ΔCt method, and each experiment was carried out in
triplicate.
Flow cytometry
For cell cycle analysis, cells were harvested by
trypsinization and fixed with 70% ethanol at 4°C overnight. The
fixed cells were rinsed twice with PBS and resuspended in propidium
iodine (PI) solution, including 50 μg/ml PI and 50 μg/ml RNaseA
(Sigma) in PBS without calcium and magnesium, and incubated at 37°C
for 30 min in the dark. The fluorescence of the cells was measured
by a FACSCalibur system (Nippon Becton Dickinson, Tokyo, Japan),
and the percentages of cells in the G1, S and G2/M phases were
determined by the ModFit program (Nippon Becton Dickinson). Flow
cytometry analysis was repeated three times with the variation in
results <20%. For analysis of >4N DNA content, cells were
harvested, fixed with ethanol and stained with PI staining buffer.
The number of cells with >4N DNA content were counted, due to
the firing of replication origins more than once per cell
cycle.
Results
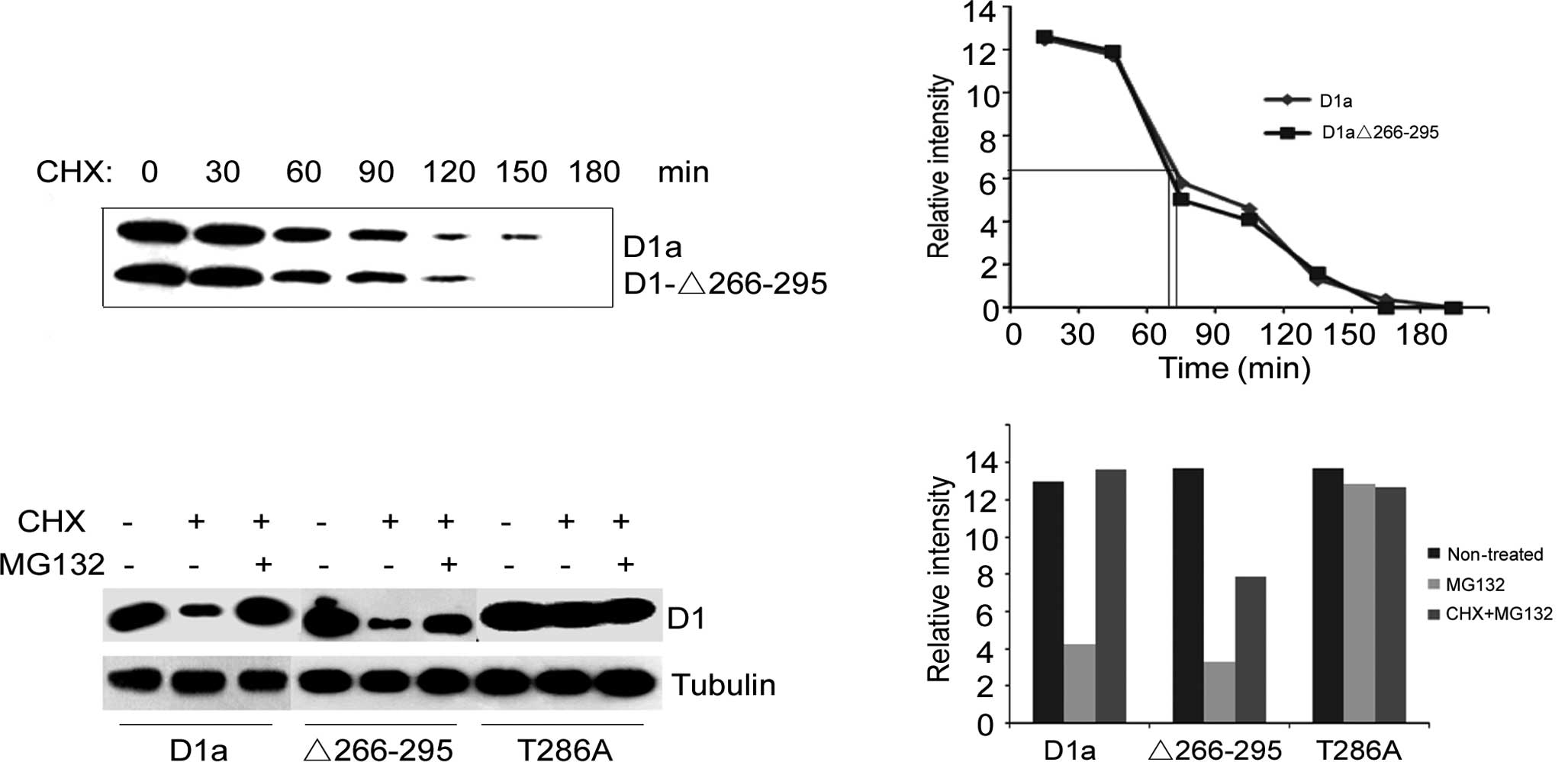
Turnover of cyclin D1-Δ266–295
Phosphorylation of Thr286 not only directs cyclin D1
nuclear export, but also promotes the rapid proteasome-dependent
destruction of cyclin D1. We therefore predicted that D1-Δ266–295
would be refractory to proteasomal degradation and exhibit an
extended half-life relative to wild-type cyclin D1. To functionally
characterize the tumor-derived cyclin D1 deletion D1-Δ266–295
protein, wild-type D1a and D1-Δ266–295 were engineered to encode a
NH2-terminal Flag-epitope tag (pFlex-D1-Δ266–295 or pFlex-D1a). The
turnover of D1-Δ266–295 vs. D1a was initially examined in NIH-3T3
cells engineered to express Flag-D1a and Flag-D1-Δ266–295
ectopically. However, inconsistent with the loss of Thr286
phosphorylation and increased nuclear retention, cyclin D1-Δ266–295
exhibited an almost identical half-life to wild-type cyclin D1
(Fig. 1A). The half-life was ~75
min for cyclin D1a and 70 min for D1-Δ266–295 (Fig. 1B). The absence of the degradation
box and Thr286 had no major effect on the turnover of cyclin
D1.
We then investigated whether the degradation of
cyclin D1-Δ266–295 resulted from proteolysis via the 26S
proteasome. To this end, specific inhibitors cycloheximide (CHX)
and MG132 were exploited to block a new protein synthesis and to
inhibit proteasome-dependent proteolysis, respectively. As shown in
Fig. 1C, the treatment of
transfected cells with CHX for 6 h resulted in a reduced level of
cyclins D1a and D1-Δ266–295 (lane 2 vs. lane 1, lane 5 vs. lane 4,
Fig. 1C). Once treated with CHX
along with MG132, the protein levels of cyclins D1a and D1-Δ266–295
in transfected cells were significantly higher than those in only
CHX-treated cells (lane 3 vs. lane 2, lane 6 vs. lane 5, Fig. 1C). As for cyclin
D1-T286A-transfected cells, treatment with CHX only, or with both
CHX and MG132, did not change the protein level of cyclin D1-T286A
(lane 7 vs. lanes 8 and 9, Fig.
1C). Taken together, these data indicate that cyclin
D1-Δ266–295 is degraded by the 26S proteasome in the same manner as
cyclin D1a, while cyclin D1-T286A is refractory. Furthermore, the
proteasome inhibitor MG132 significantly increased the cyclin D1a
protein level compared to that of cyclin D1-Δ266–295 (Fig. 1D), suggesting that there is another
degradation mechanism for cyclin D1-Δ266–295.
Subcellular localization of cyclin
D1-Δ266–295
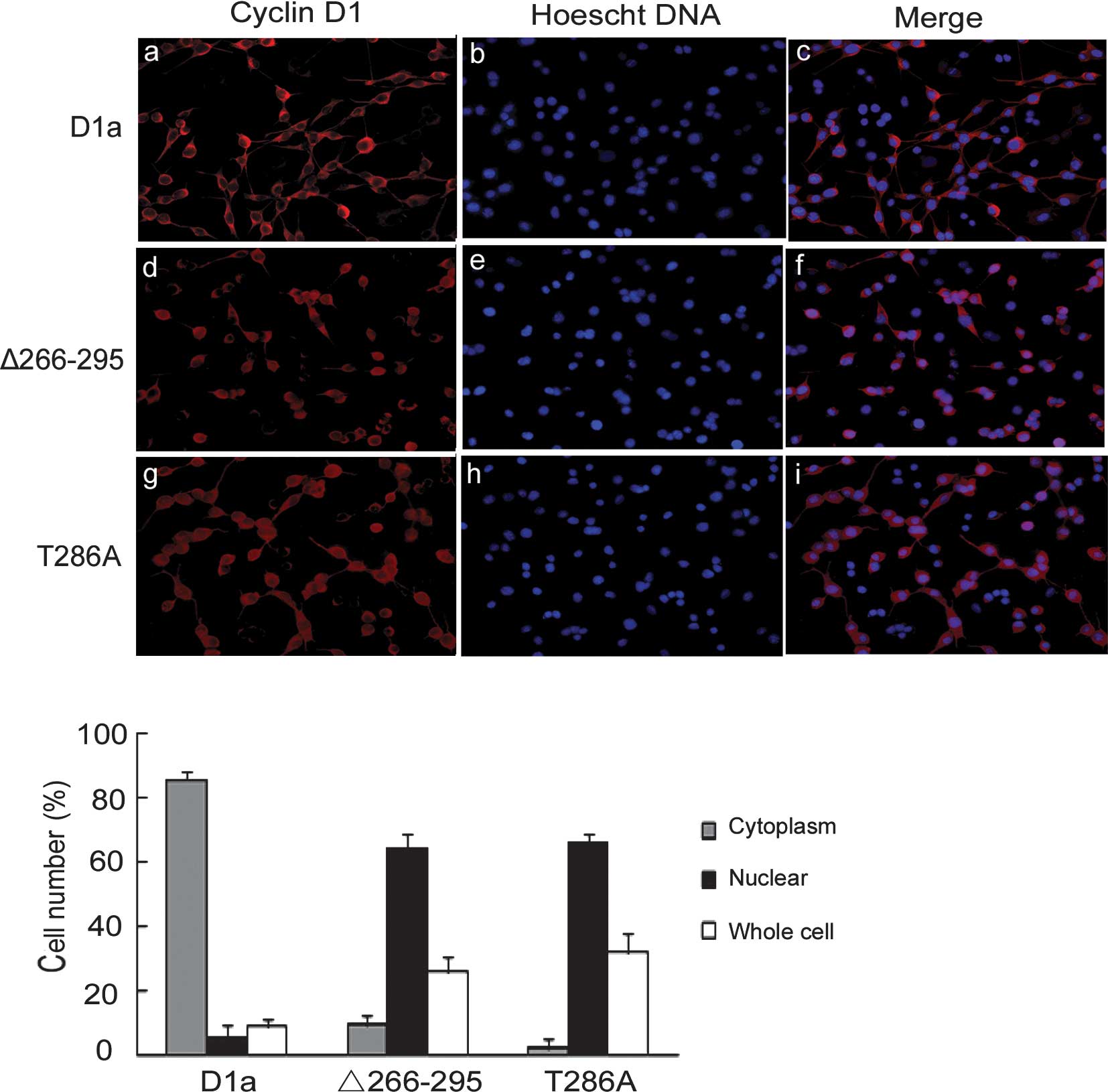
Since cyclin D1-Δ266–295 lacks the GSK-3β
phosphorylation site, we reasoned that it may be refractory to the
nuclear export directed by GSK-3β and CRM1, and exhibit a nuclear
localization pattern. To test this hypothesis, D1-Δ266–295 and D1a
localization was examined by indirect cyto-immunofluorescence
staining in asynchronous NIH3T3 cells. Staining with the M2
antibody showed an apparent cytoplasmic localization of cyclin D1a
with little nuclear overlap as expected (Fig. 2A, a-c). By contrast, cyclin
D1-Δ266–295 exhibited exclusively nuclear localization patterns
(Fig. 2A, d-f), behaving in the
same manner as the previously reported D1-T286A mutant, remaining
nuclear throughout the interphase (Fig.
2A, g-i). Quantification showed that D1a was distributed
predominantly in the cytoplasm and that only 5% of cells were
nuclear- positive, whereas the distribution of D1-Δ266–295 and
D1-T286A was predominantly nuclear at 63 and 66%, respectively
(Fig. 2B).
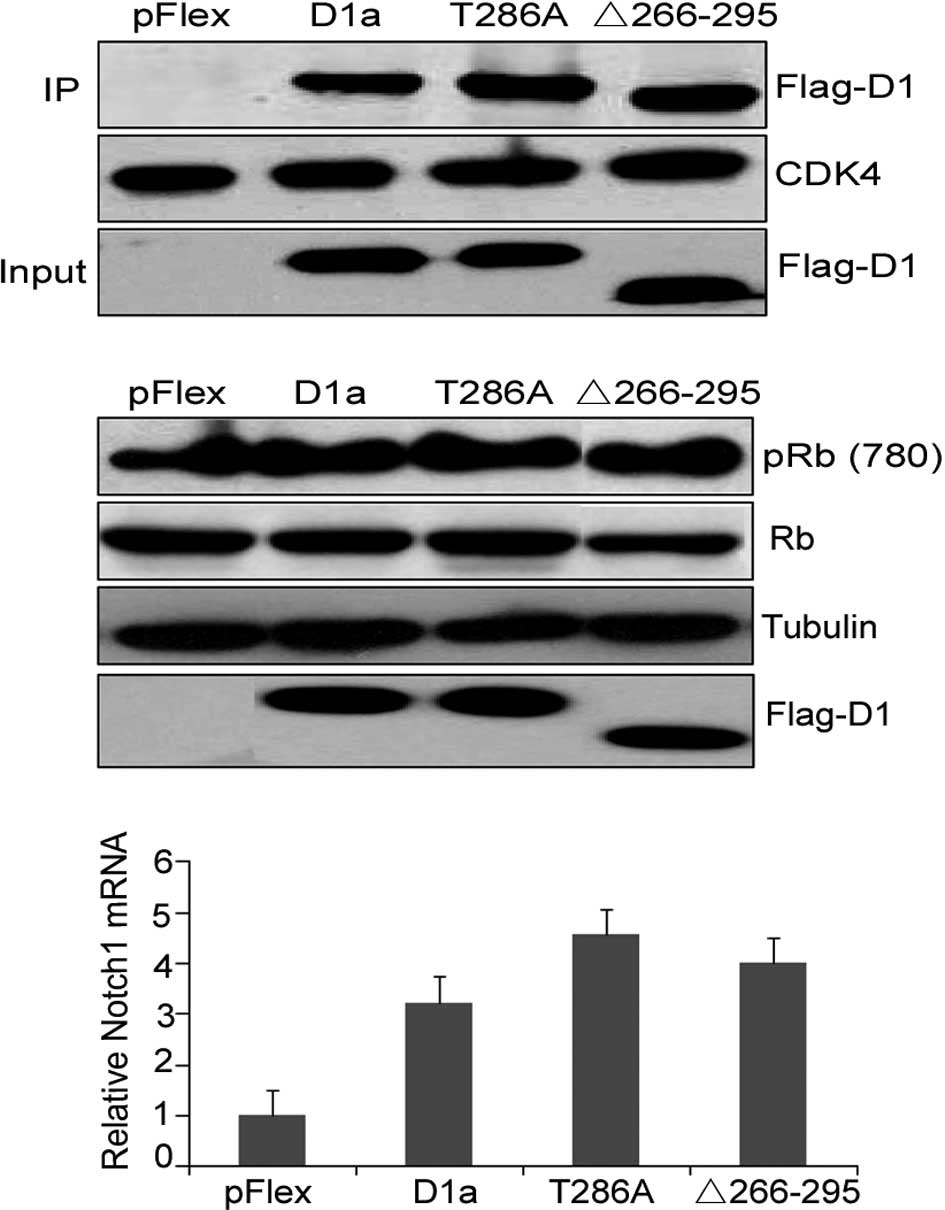
Analysis of the cyclin D1-Δ266–295/CDK4
complex formation and cyclin D1-Δ266–295 transcriptional
function
pRb is the critical target of the cyclin D1/CDK4
complex, and the cell cycle transition from the G1- to S-phase
requires the temporal activation of cyclin D1/CDK4 and subsequent
Rb phosphorylation. To investigate the ability of cyclin
D1-Δ266–295 to form binary complexes with CDK4,
co-immunoprecipitation experiments were performed in NIH-3T3 cells,
and cyclin D1/CDK4 complexes were isolated from whole cell lysates
using CDK4-specific antibody. The presence of D1a, D1-Δ266–295 and
D1-T286A was determined by immunoblotting with the M2 antibody. As
shown in Fig. 3A, D1-Δ266–295
retained the ability to bind to CDK4 to the same extent as D1a and
D1-T286A, as CDK4 was expressed at the same levels in each
precipitate (Fig. 3A, middle). A
similar expression of the various types of cyclin D1 in NIH-3T3
cells was confirmed with total protein extracts (input, lanes 2 and
3 vs. lane 4). To evaluate the ability of CDK4/D1-Δ266–295
complexes to mediate Rb phosphorylation, lysates prepared from
NIH-3T3 cells transfected with cyclin D1 mutations were subjected
to Western blot analysis with the antibody specific for
phosphor-serine at amino acid 780 (pRb780Ser), which is a site of
CDK4-mediated phosphorylation on Rb. Flag-D1a and Flag-D1-Δ266–295
efficiently promoted Rb780Ser phosphorylation when overexpressed in
NIH-3T3 cells (Fig. 3B, top; lane 1
vs. lanes 2 and 3).
Cyclin D1 also acts as a transcriptional modulator
by regulating the activity of several transcription factors. Cyclin
D1 has been shown to bind the upstream regulatory region of the
Notch1 gene where it serves to recruit CBP histone
acetyltransferase, and increases Notch1 mRNA levels in vivo.
We then determined whether D1-Δ266–295 mutation disrupts this
transcriptional ability by assessing the mRNA expression of the
Notch1 gene in the KYSE510 human esophageal carcinoma cells
overexpressing the respective cyclin D1 mutant. Real-time PCR
analysis results showed that D1-Δ266–295 increased Notch1 mRNA
levels in a similar manner to the wild-type cyclin D1 and T286A
mutations, demonstrating that the transcriptional ability of cyclin
D1-Δ266–295 was not eradicated (Fig.
3C). These results emphasized the retained structural and
functional integrity of D1-Δ266–295.
Effects of cyclin D1-Δ266–295 on cell
proliferation
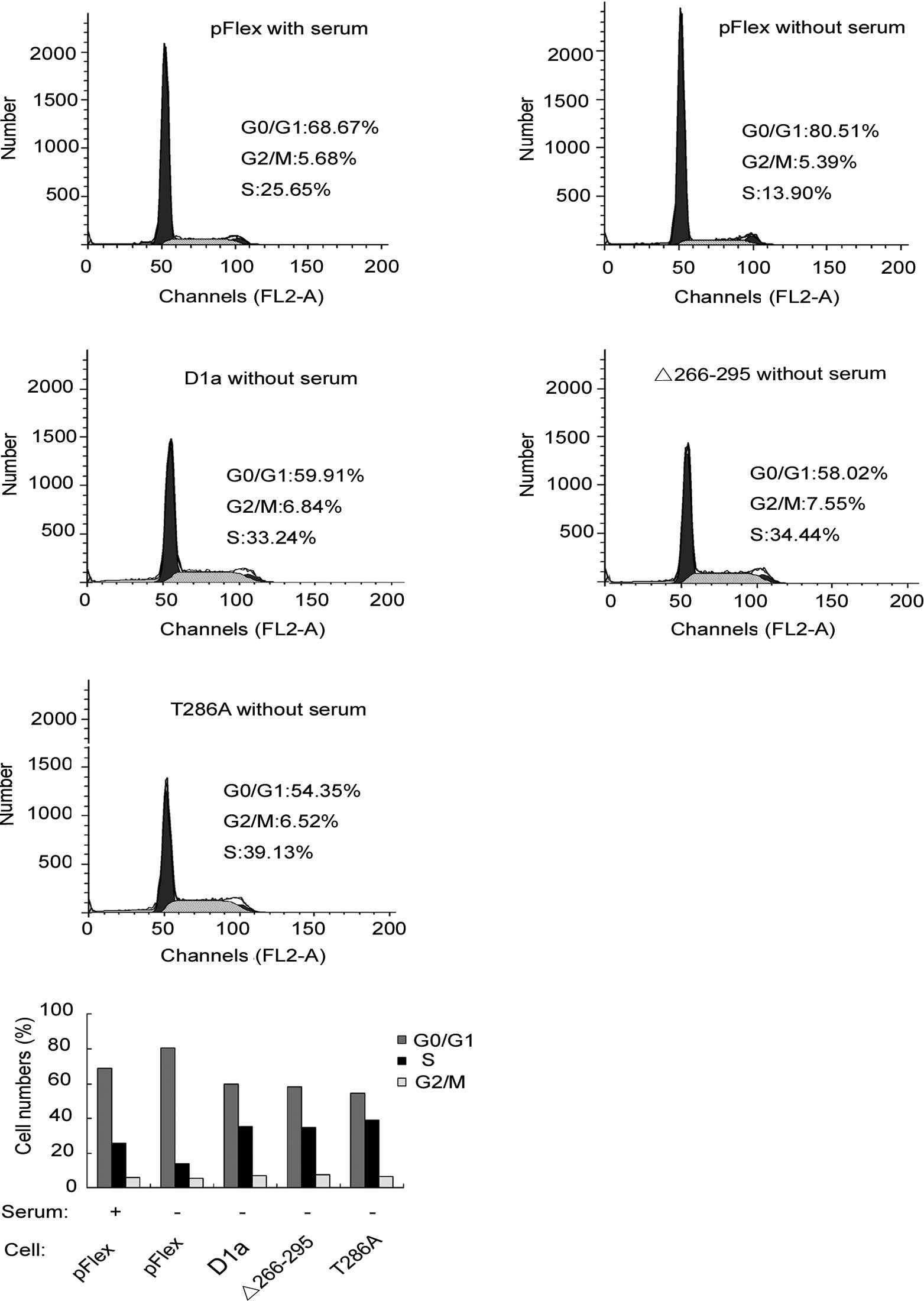
The effects of D1-Δ266–295 on cell proliferation
were investigated and compared to those of cyclins D1a and
D1-T286A. To investigate the effects of cyclin D1 on cell cycle
progression, the human esophageal carcinoma KYSE510 cells
transfected with pFlex (control), pFlex-D1a, pFlex-D1-T286A or
pFlex-D1-Δ266–295, were treated with serum starvation for 24 h.
Cells were then harvested and analyzed by flow cytometry. As shown
in Fig. 4A and B, with serum, the
fractions of cells in the G0/G1, S or G2/M phases were 68.67, 25.65
and 5.68%, respectively, for Flex-KYSE510 (control). Following 24 h
of serum deprivation, the corresponding fractions yielded were
80.51, 13.9 and 5.59%, respectively, indicating that serum
starvation blocked cells in the G1 phase as demonstrated by the
increase in the G0/G1 cell fraction (80.51 vs. 68.67%). In
comparison to Flex-KYSE510 cells, even following 24 h of serum
starvation, only 58.02% of D1-Δ266–295-KYSE510 cells were observed
to be in the G0/G1 phase, whereas as many as 34.04% were in the S
phase, a similar level to that observed with Flex-KYSE510 control
with serum. This finding suggested that D1-Δ266–295 stimulates the
KYSE510 cell cycle progression and accelerates the entry and
proportion of cells in the S phase, even without serum stimulation.
As expected, D1a-KYSE510 and D1-T286A-KYSE510 cells exhibited
similar results to those of D1-Δ266–295-KYSE510, demonstrating that
there is no difference in the promotion of cell cycle progression
between cyclins D1-Δ266–295 and D1a, although D1-Δ266–295 is
defective for the C-terminal sequences. To expand our analysis, we
also examined the effect of the exogenous expression of D1-Δ266–295
on the cell cycle distribution of HepG2 cells, a type of hepatic
tumor cell line, and similar results as those with KYSE510 were
observed (data not shown).
Effects of cyclin D1-Δ266–295 on DNA
re-replication
The increased oncogenicity of constitutively nuclear
cyclin D1T286A and transcription isoform cyclin D1b relative to
wild-type cyclin D1a suggests that nuclear retention during the S
phase is a gain-of-function characteristic. D1-Δ266–295 also
accumulated predominantly in the nucleus of expressing cells.
Therefore, we tested whether the D1-Δ266–295/CDK4 kinase would
induce the accumulation of cells harboring >4N DNA content as a
marker of DNA re-replication. Initially, we assessed the ability of
cyclin D1-Δ266–295/CDK4 complexes to drive DNA re-replication in
cooperation with Cdt1. HepG2 cells were utilized due to their high
efficacy of transfection. Consistent with our hypothesis, the
coexpression of Cdt1 along with cyclin D1-Δ266–295 resulted in a
significant increase in the >4N population, similar to D1T286A
(Fig. 5A). Cells engineered to
overexpress D1-Δ266–295 exhibited >18% of cells with >4N DNA
content, and D1T286A exhibited >26% of cells with >4N,
whereas wild-type cyclin D1a in cooperation with Cdt1 only induced
a marginal increase in the accumulation of >4N cells compared to
the control cells transfected with an empty vector (12.86 vs.
8.67%). Thus, as in the case of D1T286A, cyclin D1-Δ266–295
cooperated with Cdt1 to promote cell DNA re-replication.
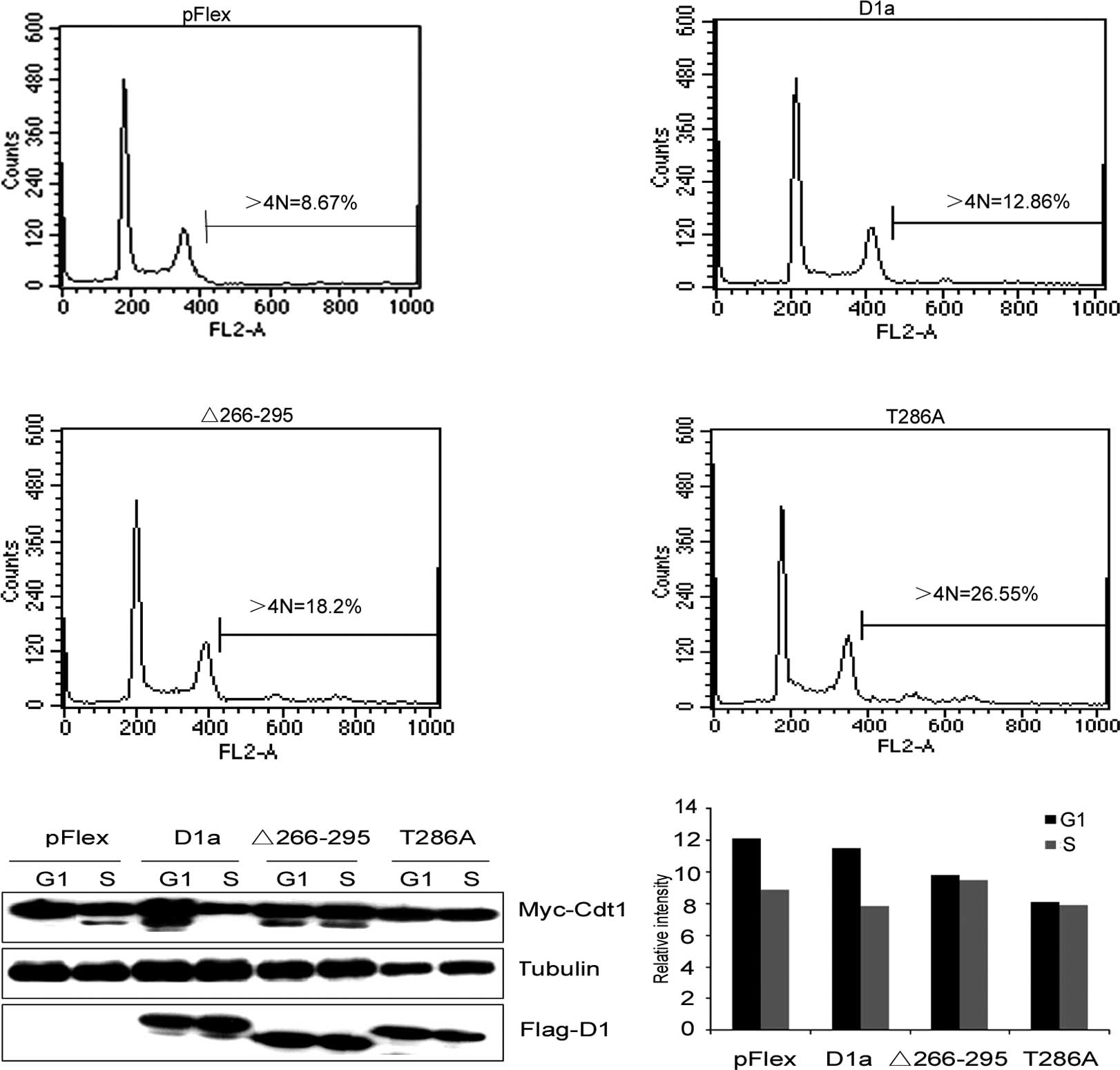 | Figure 5Cdt1, in cooperation with the
D1-Δ266–295-dependent kinase, induces DNA re-replication. (A) HepG2
cells transfected with Cdt1, along with CDK4 and cyclin D1
expression vectors, where indicated, were treated with nocodazole
for 14 h, after which cells were shaken off and replated in two
plates. The first plate, in complete media, was harvested for 6 h
after release (G1 phase). In the second dish, HU was added and
cells were harvested for 15 h after the shake-off (S phase),
stained with propidium iodide and analyzed by flow cytometry. (B)
HepG2 cells were transfected with the indicated plasmids and
synchronized in the G1 or S phase. Lysates prepared from these
cells were analyzed by Western blotting with antibodies directed
towards the proteins indicated to the right. (C) The expression
levels of Cdt1 analyzed by Quantity One software shown in (B). |
To further examine the correlation between the
nuclear retention of cyclin D1-Δ266–295/CDK4 and Cdt1 accumulation,
we determined whether the acute expression of nuclear cyclin
D1-Δ266–295 was capable of suppressing Cdt1 proteolysis. HepG2
cells were transfected with plasmids encoding myc-tagged Cdt1 along
with either wild-type or mutant cyclin D1. Cdt1 levels were
evaluated in the G1 phase (Cdt1 stable) or the S phase (Cdt1
unstable) by Western blot analysis. As shown in Fig. 5B and C, Cdt1 levels were markedly
reduced in the S phase in control cells (Fig. 5B, lanes 1 and 2), indicating that
the expression of wild-type cyclin D1a did not attenuate Cdt1
degradation (Fig. 5B, lanes 3 and
4). On the other hand, the expression of either D1-Δ266–295 or
D1T286A inhibited S phase-specific Cdt1 loss (Fig. 5B, lanes 5–8), which was consistent
with their inability to promote cell DNA re-replication.
Discussion
Cyclin D1, encoded by the CCND1 gene located on
11q13, plays a significant role in the progression of the cell
cycle (27). Cyclin D1 is known to
be frequently overexpressed in 40–60% esophageal cancers, while the
frequency of genetic alterations that directly involve the cyclin
D1 locus in esophageal cancers is only 10% (28,29).
Thus, predicting frequent alterations in pathways that regulate
cyclin D1 protein degradation is significant. Consistent with this
postulation, a number of mutations that impair the
phosphorylation-dependent nuclear export of cyclin D1 have been
identified in esophageal carcinoma and esophageal cancer-derived
cell lines (25). Similarly, the
cyclin D1 gene in endometrial cancer also possesses mutations or
deletions that are expected to affect Thr286 phosphorylation and
CRM1 binding (26). These results
indicate that mutations promoting constitutive cyclin D1 nuclear
localization are likely to be the key oncogenic events.
Cyclin D1 is a short-lived protein. Cyclin D1 is
synthesized early in the G1 phase, in response to mitogenic
signals, and is then exported from the nucleus and degraded in the
cytoplasm during the S phase, this degradation being required for
cell cycle progression (20). The
tumor-derived cyclin D1 mutation D1-Δ266–295 deleted codons from
266 to 295, the critical COOH-terminal regulatory sequences
required for cyclin D1 nuclear export. Thus, carboxyl terminal
truncated cyclin D1 protein was expected to be a constitutively
nuclear cyclin D1 and more stable than cyclin D1a. D1-Δ266–295 was
in fact found to be a constitutively nuclear localized protein.
This finding reflects the fact that D1-Δ266–295 lacks the
COOH-terminal sequences targeted by GSK-3β and CRM1. However, we
did not find any increase in the half-life of D1-Δ266–295
indicative of reduced proteolysis. Cyclins D1-Δ266–295 and D1a have
similar rates of protein turnover when expressed in the NIH3T3 cell
line. In contrast to another mutant cyclin D1-T286A which
demonstrated a 5-fold increase in the measured cyclin D1 half-life
(21), nuclear localization of
D1-Δ266–295 does not arrest its degradation. These data suggested
that cyclin D1-Δ266–295 should be more susceptible to nuclear
degradation than cyclin D1-T286A.
Similar to our observation on D1-Δ266–295, cyclin
D1b, a naturally occurring alternative splice variant of cyclin D1
lacking the fifth exon, has already been shown to be no more stable
than cyclin D1a (24,30). In addition, recent findings have
examined the role of GSK-3β in mediating cyclin D1 degradation. Guo
et al (31,32) confirmed the role of Thr286
phosphorylation in mediating cyclin D1 degradation in the S phase.
However, suppressing GSK-3β activity did not have any impact on
cyclin D1 phosphorylation or protein levels during the cell cycle.
Similarly, GSK-3β localization was not observed to alter with cell
cycle progression in MCF-7 breast cancer cells, and inhibition of
GSK-3β activity did not completely eradicate cyclin D1 degradation
(33). Since cyclin D1 mutants
lacking Thr286 remained susceptible to ubiquitination and
degradation, our data strongly suggested the existence of a second
pathway, which does not require the phosphorylation of Thr286. It
is more likely, as previously shown, that the N-terminus, but not
the C-terminus, altered cyclin D1 degradation via this pathway
(34,35).
Cyclin D1 combines with CDK4 at the cyclin box motif
and forms an active complex (36).
This complex enters the nucleus and phosphorylates Rb, promoting
the release of E2F transcription factors and thus progression from
the G1 to S phase. The cyclin box required for CDK4 interaction is
unaffected in D1-Δ266–295 and, as expected, D1-Δ266–295 bound to
CDK4 and exhibited pRb phosphorylation activity in vivo, in
the same manner as the wild-type cyclin D1. In addition,
D1-Δ266–295 retained the transcriptional function on Notch1 gene
transcription. Further investigation showed that there was no
difference in the promotion of cell cycle progression between
cyclins D1-Δ266–295 and D1a, although D1-Δ266–295 is defective for
the phosphorylation of Thr286 residue in the C-terminal region.
Cyclin D1 overexpression was reportedly not sufficient to drive
neoplastic growth, while the overexpression of the mutant cyclin
D1-T286A induced cell transformation in cell culture and triggered
B-cell lymphoma in a mouse model (17,20).
Furthermore, transgenic mice that overexpress D1T286A developed
mammary adenocarcinoma with a shorter latency relative to mice
overexpressing the wild-type cyclin D1 (37). These observations demonstrate that
subcellular localization and stabilization of cyclin D1 may exert
more profound effects on tumorigenesis than its overexpression.
This study provides evidence that cyclin D1-Δ266–295 may possess
oncogenic activity and drive neoplastic growth. This finding
suggests that in addition to the well-described G1 functions of
cyclin D1 in growth factor signaling and G1- to S-phase
progression, the constitutive nuclear retention of mutant cyclin D1
may have additional mechanisms throughout the cell cycle that
promote cell transformation.
DNA replication is a highly regulated process that
involves numerous licensing and replication factors that cooperate
to faithfully replicate DNA during each cell cycle. Loss of proper
licensing control results in deregulated DNA replication, including
DNA re-replication, which causes genome instability and
tumorigenesis (38). Previous
studies have shown that inappropriate localization of active cyclin
D1/CDK4 complex interferes with the temporal regulation of DNA
replication, contributing to genomic instability and neoplastic
transformation (39). Nuclear
accumulation of the catalytically active mutant cyclin D1T286A/CDK4
complex has been proven to stabilize Cdt1, an origin-licensing
factor that is usually degraded during the S phase to arrest
reloading of the replicative MCM helicase. Consequently, stabilized
Cdt1 continually primes DNA re-replication during the S phase and
induces genomic instability characterized by aneuploidy (39). Consistent with this finding, data
from the present study showed that the tumor-derived D1-Δ266–295
mutation triggered Cdt1 stabilization during the S phase in cell
culture, and induced a greater accumulation of >4N cells than
wild-type cyclin D1. Nuclear D1-Δ266–295, but not wild-type cyclin
D1, is capable of inhibiting Cdt1 proteolysis and promoting
re-replication, which is consistent with a previously published
study whose findings indicated that overexpressed wild-type cyclin
D1 does not induce a DNA damage response (40,41).
The above observations suggest that the genomic instability
triggered by nuclear retention of the active cyclin D1/CDK complex
is a crucial determinant to elicit the oncogenicity of cyclin D1 in
addition to its prevalent overexpression in cancers. However, the
underlying molecular mechanism remains to be determined.
In conclusion, the results provided by the present
study suggest that the features of constitutive nuclear
localization of this tumor-derived cyclin D1-Δ266–295 mutant,
contribute to the genesis and progression of neoplastic growth.
Results of the present study are likely to expand knowledge of the
oncogenicity of constitutively active cyclin D1 mutant proteins.
However, further investigation into the role played by the nuclear
cyclin D1/CDK complex in the context of genomic instability and
neoplastic transformation is required.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 30771099), and the
National S and T Major Project for Infectious Diseases
(2009ZX10004-903).
References
|
1
|
Kato J, Matsushime H, Hiebert SW, Ewen ME
and Sherr CJ: Direct binding of cyclin D to the retinoblastoma gene
product (pRb) and pRb phosphorylation by the cyclin D-dependent
kinase CDK4. Genes Dev. 7:331–342. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998.PubMed/NCBI
|
|
3
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coqueret O: Linking cyclins to
transcriptional control. Gene. 299:35–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bienvenu F, Jirawatnotai S, Elias JE, et
al: Transcriptional role of cyclin D1 in development revealed by a
genetic-proteomic screen. Nature. 463:374–378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fu M, Rao M, Bouras T, et al: Cyclin D1
inhibits peroxisome proliferator-activated receptor gamma-mediated
adipogenesis through histone deacetylase recruitment. J Biol Chem.
280:16934–16941. 2005. View Article : Google Scholar
|
|
7
|
Gladden AB and Diehl JA: Location,
location, location: the role of cyclin D1 nuclear localization in
cancer. J Cell Biochem. 96:906–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deshpande A, Sicinski P and Hinds PW:
Cyclins and cdks in development and cancer: a perspective.
Oncogene. 24:2909–2915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gillett C, Smith P, Gregory W, et al:
Cyclin D1 and prognosis in human breast cancer. Int J Cancer.
69:92–99. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sicinski P, Donaher JL, Parker SB, et al:
Cyclin D1 provides a link between development and oncogenesis in
the retina and breast. Cell. 82:621–630. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartkova J, Lukas J, Strauss M and Bartek
J: The PRAD-1/cyclin D1 oncogene product accumulates aberrantly in
a subset of colorectal carcinomas. Int J Cancer. 58:568–573. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartkova J, Lukas J, Muller H, Strauss M,
Gusterson B and Bartek J: Abnormal patterns of D-type cyclin
expression and G1 regulation in human head and neck cancer. Cancer
Res. 55:949–956. 1995.PubMed/NCBI
|
|
13
|
Hibberts NA, Simpson DJ, Bicknell JE, et
al: Analysis of cyclin D1 (CCND1) allelic imbalance and
overexpression in sporadic human pituitary tumors. Clin Cancer Res.
5:2133–2139. 1999.PubMed/NCBI
|
|
14
|
Hemmer S, Wasenius VM, Haglund C, et al:
Deletion of 11q23 and cyclin D1 overexpression are frequent
aberrations in parathyroid adenomas. Am J Pathol. 158:1355–1362.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikeguchi M, Sakatani T, Ueta T and Kaibara
N: Cyclin D1 expression and retinoblastoma gene protein (pRB)
expression in esophageal squamous cell carcinoma. J Cancer Res Clin
Oncol. 127:531–536. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin M, Inoue S, Umemura T, et al: Cyclin
D1, p16 and retinoblastoma gene product expression as a predictor
for prognosis in non-small cell lung cancer at stages I and II.
Lung Cancer. 34:207–218. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alt JR, Cleveland JL, Hannink M and Diehl
JA: Phosphorylation-dependent regulation of cyclin D1 nuclear
export and cyclin D1-dependent cellular transformation. Genes Dev.
14:3102–3114. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bodrug SE, Warner BJ, Bath ML, Lindeman
GJ, Harris AW and Adams JM: Cyclin D1 transgene impedes lymphocyte
maturation and collaborates in lymphomagenesis with the myc gene.
EMBO J. 13:2124–2130. 1994.PubMed/NCBI
|
|
19
|
Lovec H, Grzeschiczek A, Kowalski MB and
Moroy T: Cyclin D1/bcl-1 cooperates with myc genes in the
generation of B-cell lymphoma in transgenic mice. EMBO J.
13:3487–3495. 1994.PubMed/NCBI
|
|
20
|
Gladden AB, Woolery R, Aggarwal P, Wasik
MA and Diehl JA: Expression of constitutively nuclear cyclin D1 in
murine lymphocytes induces B-cell lymphoma. Oncogene. 25:998–1007.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Diehl JA, Zindy F and Sherr CJ: Inhibition
of cyclin D1 phosphorylation on threonine-286 prevents its rapid
degradation via the ubiquitin-proteasome pathway. Genes Dev.
11:957–972. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Germain D, Russell A, Thompson A and
Hendley J: Ubiquitination of free cyclin D1 is independent of
phosphorylation on threonine 286. J Biol Chem. 275:12074–12079.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis
and subcellular localization. Genes Dev. 12:3499–3511. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu F, Gladden AB and Diehl JA: An
alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear
oncogene. Cancer Res. 63:7056–7061. 2003.PubMed/NCBI
|
|
25
|
Benzeno S, Lu F, Guo M, et al:
Identification of mutations that disrupt phosphorylation-dependent
nuclear export of cyclin D1. Oncogene. 25:6291–6303. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moreno-Bueno G, Rodriguez-Perales S,
Sanchez-Estevez C, et al: Cyclin D1 gene (CCND1) mutations in
endometrial cancer. Oncogene. 22:6115–6118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cobrinik D: Pocket proteins and cell cycle
control. Oncogene. 24:2796–2809. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shinozaki H, Ozawa S, Ando N, et al:
Cyclin D1 amplification as a new predictive classification for
squamous cell carcinoma of the esophagus, adding gene information.
Clin Cancer Res. 2:1155–1161. 1996.PubMed/NCBI
|
|
29
|
Shamma A, Doki Y, Shiozaki H, et al:
Cyclin D1 overexpression in esophageal dysplasia: A possible
biomarker for carcinogenesis of esophageal squamous cell carcinoma.
Int J Oncol. 16:261–266. 2000.PubMed/NCBI
|
|
30
|
Leveque C, Marsaud V, Renoir JM and Sola
B: Alternative cyclin D1 forms a and b have different biological
functions in the cell cycle of B lymphocytes. Exp Cell Res.
313:2719–2729. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo Y, Yang K, Harwalkar J, et al:
Phosphorylation of cyclin D1 at Thr 286 during S phase leads to its
proteasomal degradation and allows efficient DNA synthesis.
Oncogene. 24:2599–2612. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang K, Guo Y, Stacey WC, et al: Glycogen
synthase kinase 3 has a limited role in cell cycle regulation of
cyclin D1 levels. BMC Cell Biol. 7:332006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alao JP, Stavropoulou AV, Lam EW, Coombes
RC and Vigushin DM: Histone deacetylase inhibitor, trichostatin A
induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast
cancer cells. Mol Cancer. 5:82006. View Article : Google Scholar
|
|
34
|
Feng Q, Sekula D, Muller R, Freemantle SJ
and Dmitrovsky E: Uncovering residues that regulate cyclin D1
proteasomal degradation. Oncogene. 26:5098–5106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Agami R and Bernards R: Distinct
initiation and maintenance mechanisms cooperate to induce G1 cell
cycle arrest in response to DNA damage. Cell. 102:55–66. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Alao JP: The regulation of cyclin D1
degradation: roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 6:242007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin DI, Lessie MD, Gladden AB, Bassing CH,
Wagner KU and Diehl JA: Disruption of cyclin D1 nuclear export and
proteolysis accelerates mammary carcinogenesis. Oncogene.
27:1231–1242. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Truong LN and Wu X: Prevention of DNA
re-replication in eukaryotic cells. J Mol Cell Biol. 3:13–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aggarwal P, Lessie MD, Lin DI, et al:
Nuclear accumulation of cyclin D1 during S phase inhibits
Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA
rereplication. Genes Dev. 21:2908–2922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spruck CH, Won KA and Reed SI: Deregulated
cyclin E induces chromosome instability. Nature. 401:297–300. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tort F, Bartkova J, Sehested M, Orntoft T,
Lukas J and Bartek J: Retinoblastoma pathway defects show
differential ability to activate the constitutive DNA damage
response in human tumorigenesis. Cancer Res. 66:10258–10263. 2006.
View Article : Google Scholar : PubMed/NCBI
|