Introduction
As the most refractory tumors of the central nervous
system, gliomas comprise 33.3–58.9% of intracranial tumors. The
high-grade gliomas in particular (e.g., glioblastoma multiforme)
have a dismal prognosis and a high recurrence rate; despite
advances in multimodality therapy, the median survival is only 1
year (1,2). In recent years, the cancer stem cells
theory (3) and the finding of
glioma stem cells (4–6) have been the highlights of glioma
research. Brain glioma stem cells (BGSCs) have been proven to be
the ‘seed’ cells of, and be responsible for, the initiation and
development of gliomas, and thus have become new targets of glioma
therapy (7). Currently the research
on BGSCs mainly focuses on cell experiments, which demand a large
number of cultured BGSCs as the first step. In this study, we
introduce a simplified and modified procedure to culture, identify
and purify BGSCs from resected samples of glioma patients, which is
much more convenient and practical for the subsequent BGSC
experiments.
Materials and methods
Tissue collection and grading
Brain glioma specimens were obtained within 30 min
of surgical resection and were processed. The pathological grade of
each specimen was confirmed by chief neuropathologists according to
World Health Organization criteria. All 17 cases were inpatients in
the Department of Neurosurgery of the First Hospital of China
Medical University, China, between October 2008 and January 2009.
The protocol of this study was approved by the Institutional Review
Boards of the First Clinical Hospital, China Medical
University.
Reagents and antibodies
Fetal bovine serum (FBS, qualified, Gibco,
Invitrogen, Carlsbad, CA, USA), Dulbecco’s modified Eagles medium
(DMEM)/F12 (Gibco), B-27 supplement (without serum and vitamin A,
Invitrogen), recombinant human epidermal growth factor (rhEGF,
Invitrogen), basic recombinant human fibroblast growth factor
(rhFGF-b, Invitrogen), mouse monoclonal anti-CD133 antibodies
(Abcam, Cambridge, UK), monoclonal rabbit anti-GFAP (glial
fibrillary acidic protein) antibodies (Bioworld, Dublin, OH, USA),
TU-20 (monoclonal mouse anti-β-tubulin III isoform, C-terminus,
Millipore, Billerica, MA, USA), Cy3-conjugated goat anti-mouse
secondary antibodies (Sigma, St. Louis, MA, USA), Cy3-conjugated
goat anti-rabbit secondary antibodies (Sigma), fluorescein
isothiocyanate (FITC)-conjugated goat anti-mouse antibodies (Sigma)
and 4,6-diamidino-2-phenylindole (DAPI, Sigma) were used. The other
common reagents were all analytically pure.
Culture of primary glioma cells and tumor
spheres
Tumor-sphere cultures were performed according to
reported procedures (4–6,8), with
minor modifications. Following resection, the tumor tissues were
washed, minced in phosphate-buffered saline (PBS) and subjected to
enzymatic dissociation. The tissues were then mechanically
dissociated with a graded series of fire-polished Pasteur pipettes,
and passed through a series of cell strainers, and centrifuged at
800 × g for 5 min. Tumor cells were resuspended in DMEM/F12
containing 15% FBS and plated at a density of 2×105 live
cells/ml. When tumor cells grew as monolayers in flasks, the medium
was changed to committed stem cell medium (serum-free DMEM/F-12,
1:50 of B-27 supplement, 20 ng/ml rhEGF, 20 ng/ml rhFGF-b, 100
IU/ml penicillin G and 100 μg/ml streptomycin). Fresh rhEGF and
rhFGF-b were added each week and the medium was changed twice a
week. Cells were maintained in a standard tissue culture incubator
with 5% CO2 and 100% relative humidity at 37°C.
Proliferation assay and limited dilution
assays
In the proliferation assay, when suspending cells
emerged and the primary tumor spheres were clear, they were
collected and dissociated into single-cell suspensions through a
fire-polished Pasteur pipette, and then reseeded in the stem cell
media at the same cell density. Each cell line was serially
passaged over 4 generations. To evaluate the self-renewal capacity
of tumor spheres, tumor spheres were mechanically dissociated into
single-cell suspensions and reseeded into 96-well microwell plates
by limited dilution at a cell density of 1–2 live cells per well.
Each well was fed with fresh stem cell media every 3–4 days. The
formation of cell clones was inspected under a phase-contrast
microscope after 7–10 days.
Immunocytochemistry assays
The tumor spheres of approximately the 4th passage
were collected and plated onto anti-peeling slides and incubated
with stem cell media for 4 h. Following firm adhesion, the tumor
spheres were fixed in 4% paraformaldehyde and incubated at 4°C with
mouse monoclonal anti-CD133 antibodies diluted at 1:200 according
to the manufacture’s instructions. Cy3-conjugated goat anti-mouse
secondary antibodies diluted at 1:50 were added 24 h later and
incubated for 2 h at room temperature.
To assess the multipotency of the tumor spheres, the
tumor spheres were harvested and subjected to a differentiation
assay by plating onto coverslips precoated with poly-L-lysine in
DMEM/F-12 media containing 15% FBS and in the absence of growth
factors or B-27 supplement. The media were changed every 2 days.
The differentiated cells were fixed 7 days later, incubated with
monoclonal rabbit ant-GFAP (1:200) for glial cells and TU-20
(1:200) for neurons overnight at 4°C. Secondary antibodies
(Cy3-conjugated goat anti-rabbit and FITC-conjugated goat
anti-mouse, 1:250) were then added and incubated for 2 h at room
temperature.
For all of the staining, the cell nuclei were
counterstained with DAPI for 5 min. In the control samples, primary
antibodies were replaced by isotype IgG. A fluorescence microscope
(Olympus BX61) was then used to observe and capture images of the
results. The excitation wavelength was 554 nm for Cy3, 488 nm for
FITC and 350 nm for DAPI.
Results
Tumor spheres culture
Tumor spheres were successfully cultured in 8 out of
all 17 cases of glioma samples, including 2 primary glioblastoma
multiformes (WHO gradeIV), 1 recurrent anaplastic oligodendroglioma
and 5 anaplastic astrocytomas (WHO gradeIII). The patients
consisted of 3 males and 5 females. The ages varied from 44–73
years old, and the average age was 53.75 years old.
The monolayers formed 24–72 h after the single-cell
suspensions were seeded in the serum-containing media. When
switched into the stem cell media, single cell division occurred
after 48–72 h, followed by the formation of large numbers of
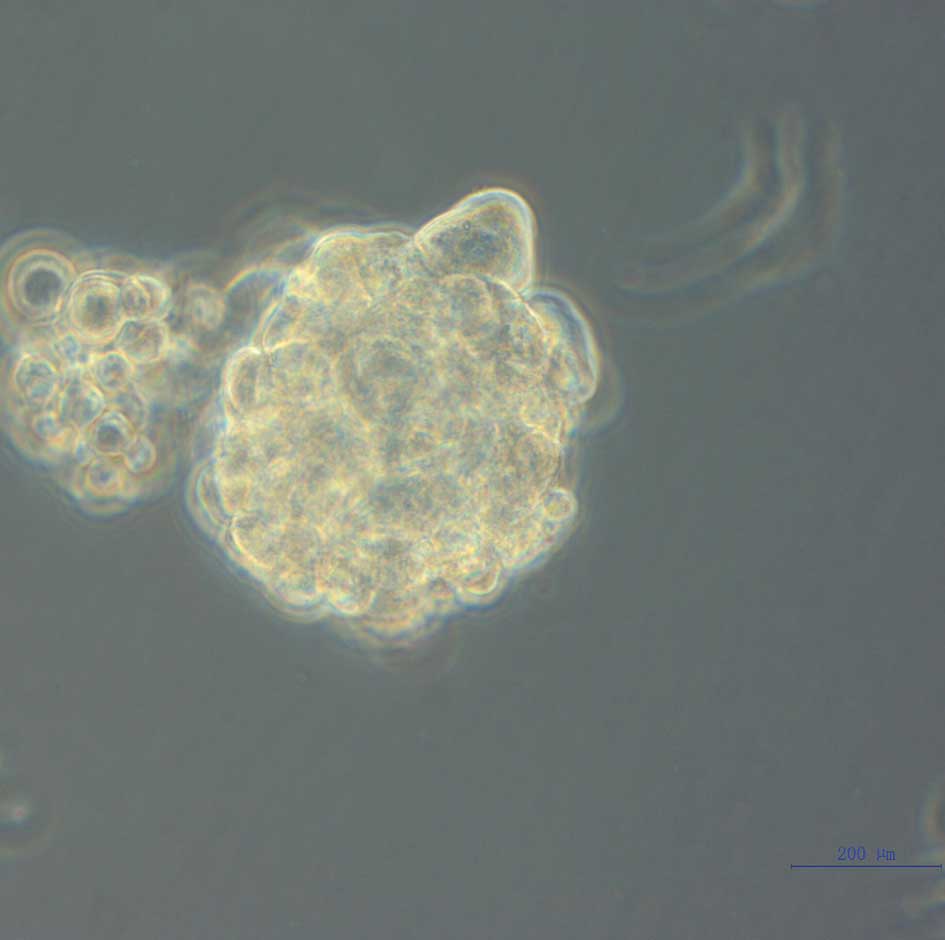
‘neurosphere-like’ tumor spheres within 5–7 days. These tumor
spheres were spherical or oval in suspending or semi-suspending
states, with fine light refraction, consisting of 4–10 cells per
sphere. Certain tumor spheres were of irregular morphology. The
growth velocity was slow in the first few weeks, but within 2 weeks
the majority of spheres had increased their diameters by 5–10-fold.
Inspected under a microscope, the tumor spheres showed a smooth
edge without prominent protrusion, and had poor light transmittance
in the center as a result of a higher cell density. The majority of
monolayer tumor cells still presented adherence, loss of
proliferation, and subsequent differentiation, while tumor spheres
remained suspended, continuing to proliferate and increase in cell
numbers. Along with the growth of tumor spheres, a few adherent
cells showed disaggregation, fragmentation, condensation and cell
pyknosis, which manifested the progress of apoptosis. After
approximately 2 weeks of culture, the formation of tumor spheres
was observed and images were captured under a phase-contrast
microscope (Fig. 1).
Proliferation and self-renewal
assessment
In the proliferation assay, tumor spheres were
dissociated into single-cell suspension and passaged at a ratio of
1:2 or 1:3. Cell cleavage occurred in 48 h and new tumor spheres
formed within 1 week. Serial passage revealed the tumor sphere
cells maintained a favorable proliferation ability after at least 4
generations. In a monoclonal formation assay, the single cells from
tumor spheres were serially diluted and reseeded in microwells.
Counted under a microscope, it demonstrated that more than 50% of
the single cells in microwells were capable of forming new
secondary tumor spheres, although the diameters were generally less
than those of the primary spheres. Each new secondary sphere
contained 10–40 cells, showing the same morphology as the parent
sphere. This assay proved that individual cells from the parent
tumor spheres were endowed with the ability to self-renew and form
new secondary sphere colonies.
Immunocytochemistry identification
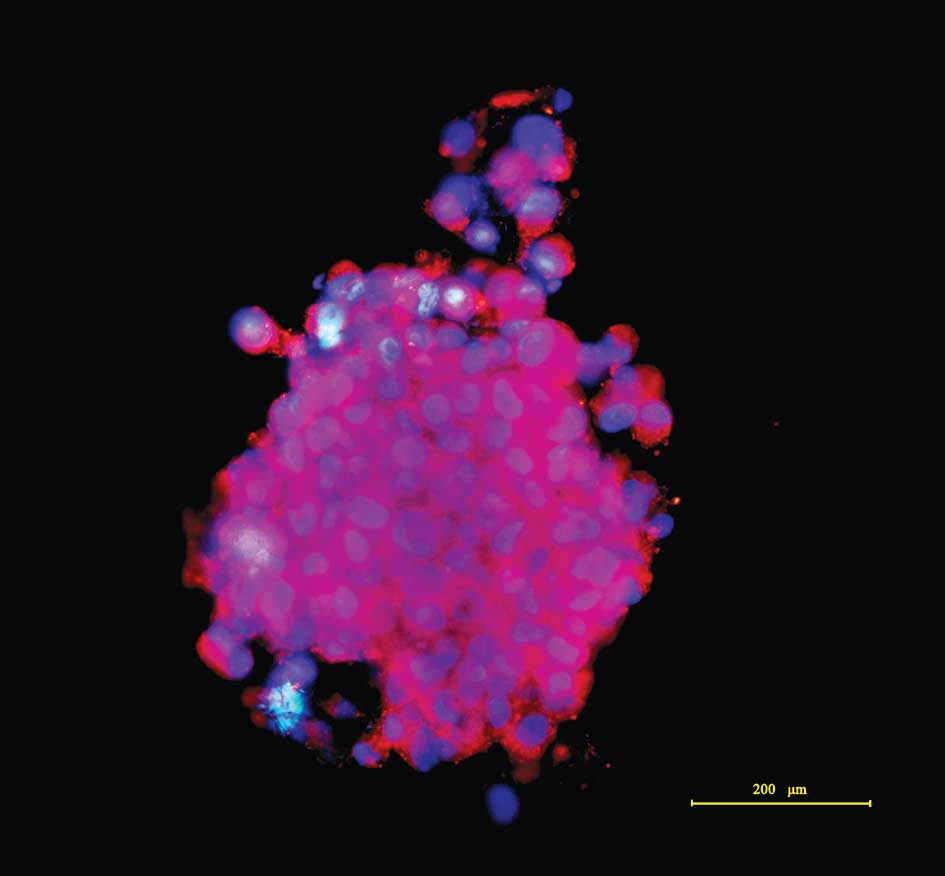
The tumor spheres were immunostained using CD133,
the committed BGSC marker (4,8–11).
After firmly attaching to the anti-peeling slides, an
immunocytochemical assay exhibited that the majority of tumor
sphere cells were CD133-positive in a plasma membrane staining
pattern under an immunofluorescence microscope. The nuclei were
counterstained, exhibiting marked nuclear atypia. Images of the
stained tumor spheres were captured, as shown in Fig. 2.
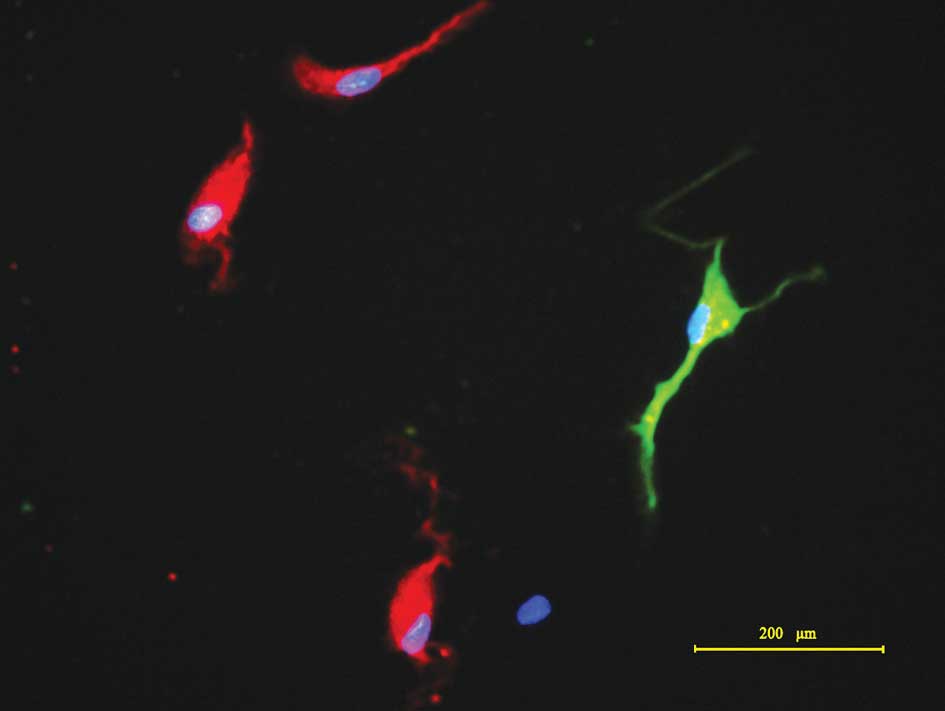
During induced differentiation, cell division in the
spheres markedly decelerated and cells attached to the substrates,
with cells migrating upon exposure to differentiation conditions.
The tumor spheres became flat, exhibiting a radial morphology. The
migrated cells formed monolayers, comprising various cell types
with marked heteromorphism. Following differentiation in media with
15% FBS for 7 days, the cells were fixed and subjected to
immunocytochemical detection. The results demonstrated that cells
differentiated from the tumor spheres were positive for β-tubulin
III and GFAP (Fig. 3), consistent
with findings from other studies (4,10,11).
The results indicated that CD133+ tumor spheres were
multipotent for at least two neural cell types, neurons and
astrocytes, which shows a multilineage differentiation ability.
Discussion
Gliomas are the most frequently diagnosed and
aggressive primary intracranial tumors, with poor outcome. The
mortality rate has remained on the increase in recent years; only
less than 5% of patients survive the first 5 years, even after
radical therapeutic strategies (12). Despite advances in glioma research
and treatment in recent decades, prognosis has not substantially
improved and the median survival period has been elevated by only
9–10 weeks (13). Thus, it is
necessary and practical to find new targets and investigate new
therapies based on glioma pathogenetic mechanisms. The theory of
BGSCs provided new targets for the cure of gliomas (4–6,11,14).
BGSCs are defined as a small population of cells capable of
extensive proliferation, self-renewal, multipotent differentiation
and tumor initiation. As the origins of glioma cells and the
critical factors, BGSCs determine glioma propagation, progression
and therapeutic resistance. Subsequently, BGSCs with a
tumor-initiating ability have been isolated from GBMs,
medulloblastomas, ependymomas and anaplastic oligoastrocytomas, and
certain biological characteristics have been investigated (4,5,8,11,15,16).
These findings suggest a hierarchical model in which glioma arises
from BGSCs and progresses through mechanisms similar to a
developmental process. It is well known that the eradication of
gliomas can only be accomplished by targeting BGSCs. Nevertheless,
little is known with regards to the biological mechanisms of
BGSCs.
CD133, a 120 kDa cell-surface protein, which is a
hallmark of normal human neural precursors, has been generally
accepted as the marker of BGSCs regardless of whether they are
solid tumors or glioma cell lines in vitro, although it is
shared by other stem cells (4,9–11,16).
In addition, some investigators used a flow cytometry-based side
population (SP) technique to isolate BGSCs based on the
characteristic that BGSCs were capable of excluding the fluorescent
dye Hoechst 33342 (17). However,
Hoechst 33342 is a type of liposoluble DNA-binding fluorescent dye
and is cytotoxic to various cell types. Therefore, it is
inappropriate to compare the respective biological features of
sorted SP cells and non-SP cells. Thus, the SP sorting technique
still calls for improvement. It is a key issue to find specific
phenotype markers for BGSCs in subsequent studies, which may
benefit further investigations on BGSCs.
The culture techniques for BGSCs are similar to the
procedures for other stem cell types, such as serum-free media
without adhesion factors and supplementation of rhEGF, rhFGF-b and
B-27 (or N2 supplementation) (4–6,15,18,19).
With regards to the sorting techniques, there are two basic methods
available, one is immunomagnetic beads or fluorescent activated
cell sorting (FACS) based on the surface marker CD133; and the
other method is the SP cell sorting technique based on a
drug-resistant gene, such as an ATP-binding cassette or a multidrug
resistance gene (MDR) (17,20). In either case, the supposed BGSCs
are sorted, then identified and cultured. The procedures are
complicated and expensive; following prolonged incubation,
labeling, sorting and rinsing, the isolated BGSCs may not retain
adequate viability for the subsequent culture process or relevant
biological experiments. In this study, we simplified the steps of
culture and identification to obtain expected BGSCs relatively
rapidly and efficiently. The detailed culture process is the same
as reported, but the isolation and identification are more
convenient as described above. The results proved that the cultured
tumor spheres could be serially passaged and possessed self-renewal
ability, expressed the preferred BGSCs marker CD133, and
differentiated into cells expressing β-tubulin III and GFAP, which
were consistent with other findings (4,6,17).
These results proved the ‘stemness’ of tumor spheres, therefore
they could be defined as CD133+ BGSCs.
Compared with conventional methods, we did not use
the sorting techniques (immunomagnetic beads, FACS, or SP
techniques) or animal experiments to initiate new glioma. As is
known, there are mainly 2 types of methods to purify BGSCs;
conditioned purification or specific marker sorting combined with
conditioned culture. Although target cells can be sorted using
specific markers, the limitations remain clear, such as the long
duration of the procedure, the high expense, complicated steps and
loss of cell viability. Since the BGSCs were cultured in suspended
tumor spheres, only the suspension cells were harvested and
serially passaged during culture. Thus, the majority of non-BGSCs
were excluded after 3–5 generations and enough target cells of high
purity were obtained. This result was confirmed by
immunocytochemical staining, as shown in Fig. 2. The fibroblasts were also excluded
during the suspension culture. When the BGSCs grew to a certain
density and number, the propagation rate accelerated and the
passage interval shortened, which were adapted for the relevant
investigations. Furthermore, the animal experiment for
tumorigenecity is currently regarded as a gold standard as it may
verify the ability of BGSCs to initiate new gliomas, although the
results are not always reliable. A number of experiments have found
that either CD133+ or CD133− glioma cells may
result in gliomas (9,19). For instance, investigators have
reported that CD133− glioma cells formed new gliomas in
nude mice (19). Certain
investigators (21,22) also stated that it is not only the
cancer stem cells that possess the ability of tumor initiation
after a series of assays of lymphoma or leukemia stem cell
xenografts. In our opinion, the NOD/SCID mice mostly selected for
tumorigenecity experiments provide a glioma growing environment
that is extremely different to the microenvironment of spontaneous
gliomas. Subsequently, the animal experiments were not always
appropriate for the identification of BGSCs and were not included
in our study, which saved time and expense.
Currently, BGSCs have attracted much attention and
have been extensively investigated in fundamental fields such as
BGSC biological characteristics, and clinical fields such as
immunotherapy or gene therapy targeting BGSCs. Cytological
experiments are substantial for BGSC research and obtaining enough
viable BGSCs for experiments is a pivotal problem. Although
numerous types of glioma cell lines were employed for BGSCs
experiments, their biological features have significantly changed
following long-term culture and serial passage in vitro and
cannot mirror the primary characteristics of gliomas. Therefore,
culturing BGSCs from clinical samples is an appropriate option.
Nevertheless, the classical procedures comprise relatively
complicated steps and a series of assays, and BGSCs finally
cultured and isolated may not retain sufficient viability and
natural properties as expected. In this study, we simplified the
procedures and successfully grew BGSCs from resected glioma samples
of clinical cases. The flow cytometry or immunomagnetic bead
sorting methods were not used; conditioned culture and serial
passage were used instead. The animal experiments for glioma
tumorigenecity were also not performed due to controversies
regarding their effectiveness. Within 3–5 generations, a
considerable number of BGSCs were harvested with a high purity,
which was confirmed by immunocytochemical identification of the
tumor spheres. The results proved that this method is time-saving,
cost-effective and convenient, contributing to the subsequent
studies on BGSCs.
Acknowledgements
This study was supported by the Foundation of
Liaoning Provincial Commission of Science Technology (No.
2007408001–5).
References
|
1
|
Hess KR, Broglio KR and Bondy ML: Adult
glioma incidence trends in the United States, 1977–2000. Cancer.
101:2293–2299. 2004.PubMed/NCBI
|
|
2
|
Reardon DA, Rich JN, Friedman HS and
Bigner DD: Recent advances in the treatment of malignant
astrocytoma. J Clin Oncol. 24:1253–1265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jordan CT, Guzman ML and Noble M: Cancer
stem cells. N Engl J Med. 355:1253–1261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galli R, Binda E, Orfanelli U, et al:
Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hemmati HD, Nakano I, Lazareff JA, et al:
Cancerous stem cells can arise from pediatric brain tumors. Proc
Natl Acad Sci USA. 100:15178–15183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.PubMed/NCBI
|
|
7
|
Tunici P, Irvin D, Liu G, et al: Brain
tumor stem cells: new targets for clinical treatments? Neurosurg
Focus. 20:272006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yi L, Zhou ZH, Ping YF, et al: Isolation
and characterization of stem cell-like precursor cells from primary
human anaplastic oligoastrocytoma. Mod Pathol. 20:1061–1068. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beier D, Hau P, Proescholdt M, et al:
CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show
differential growth characteristics and molecular profiles. Cancer
Res. 67:4010–4015. 2007.
|
|
10
|
Huang Q, Dong J, Zhu YD, et al: Isolation
and culture of tumor stem cells from human brain glioma tissues.
Zhonghua Zhong Liu Za Zhi. 28:331–333. 2006.PubMed/NCBI
|
|
11
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Claus EB and Black PM: Survival rates and
patterns of care for patients diagnosed with supratentorial
low-grade gliomas: data from the SEER program, 1973–2001. Cancer.
106:1358–1363. 2006.PubMed/NCBI
|
|
13
|
Davis FG, Freels S, Grutsch J, Barlas S
and Brem S: Survival rates in patients with primary malignant brain
tumors stratified by patient age and tumor histological type: an
analysis based on Surveillance, Epidemiology, and End Results
(SEER) data, 1973–1991. J Neurosurg. 88:1–10. 1998.PubMed/NCBI
|
|
14
|
Clarke MF, Dick JE, Dirks PB, et al:
Cancer stem cells–perspectives on current status and future
directions: AACR Workshop on cancer stem cells. Cancer Res.
66:9339–9344. 2006.
|
|
15
|
Taylor MD, Poppleton H, Fuller C, et al:
Radial glia cells are candidate stem cells of ependymoma. Cancer
Cell. 8:323–335. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan X, Curtin J, Xiong Y, et al:
Isolation of cancer stem cells from adult glioblastoma multiforme.
Oncogene. 23:9392–9400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kondo T, Setoguchi T and Taga T:
Persistence of a small subpopulation of cancer stem-like cells in
the C6 glioma cell line. Proc Natl Acad Sci USA. 101:781–786. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee J, Kotliarova S, Kotliarov Y, et al:
Tumor stem cells derived from glioblastomas cultured in bFGF and
EGF more closely mirror the phenotype and genotype of primary
tumors than do serum-cultured cell lines. Cancer Cell. 9:391–403.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Sakariassen PO, Tsinkalovsky O, et
al: CD133 negative glioma cells form tumors in nude rats and give
rise to CD133 positive cells. Int J Cancer. 122:761–768. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chua C, Zaiden N, Chong KH, et al:
Characterization of a side population of astrocytoma cells in
response to temozolomide. J Neurosurg. 109:856–866. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kelly PN, Dakic A, Adams JM, Nutt SL and
Strasser A: Tumor growth need not be driven by rare cancer stem
cells. Science. 317:3372007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kennedy JA, Barabe F, Poeppl AG, Wang JC
and Dick JE: Comment on ‘Tumor growth need not be driven by rare
cancer stem cells’. Science. 318:17222007.
|