Introduction
Breast cancer has a high incidence worldwide
(1). Factors that may play a role
in the onset and progression of breast cancer include genetic,
metabolic and lifestyle-associated risk factors. Obesity is a risk
factor via its role in increasing circulating estrogen, insulin,
insulin-like growth factor and adipokines, all of which may
contribute to cancer initiation and progression (2). Mutations in certain tumour suppressor
genes also contribute to risk of cancer (3). One such tumour suppressor is LKB1,
which is found to be mutated in Peutz-Jegher syndrome and a number
of other types of cancer, including certain types of breast cancer
(4). LKB1 is a serine/threonine
kinase that phosphorylates downstream substrates, such as
AMP-activated protein kinase (AMPK) and related kinases (5). AMPK is a master metabolic regulator of
energy in all cells and structurally is a trimeric enzyme
comprising 3 subunits, α, β, and γ. The catalytic activity belongs
to the α subunit, while the regulatory functions belong to the
other two subunits. AMPK is activated in response to
phosphorylation of the critical amino acid residue Thr172. This
phosphorylation process is catalysed by a number of upstream
kinases including LKB1 and calmodulin-dependent kinase kinase
(CaMKK) (6).
The ability of LKB1 to phosphorylate and activate
AMPK, and, moreover, the observation that absence of LKB1 in
certain cell types is accompanied by a lack of AMPK activity,
indicates that the action of LKB1 as a tumour suppressor is
attributed, at least in part, to AMPK (5). This assessment was supported and
consolidated by a number of observations. AMPK was reported to
activate and phosphorylate the trans-active domain of p53 (7) and p21 (8), disrupting the cell cycle. AMPK has
also been shown to inhibit mTOR and, thereby, protein biosynthesis.
AMPK enhances the formation of the tuberous sclerosis complex 1
(TSC1)/tuberous sclerosis complex 2 (TSC2) complex which
dephosphorylates and inactivates Rheb, an upstream activator of
mTOR (9). Lipogenic enzymes, fatty
acid synthase (FA) and acetyl CoA carboxylase (ACC), are highly
expressed in cancer cells as a result of the high demand for fatty
acids that are required to be incorporated in the plasma membranes
of the newly dividing cells. The two enzymes were suppressed by
AMPK activation in cancer cells (10).
There have been reports of apparent anti-cancer
effects of AMPK activators in breast cancer cells (11). The present study consolidates and
expands on these observations and describes the results of
investigations on the effect of AMPK activation on cell
proliferation and apoptosis in three breast cancer cell types
differing in their estrogen receptor (ER) and p53 status.
Materials and methods
Materials
5-aminoimidazole carboxamide ribonucleotide (AICAR)
and phenformin were obtained from Sigma-Aldrich (Poole, Dorset,
UK). Dulbecco’s minimal essential medium (DMEM) was obtained from
Gibco BRL (Paisley, UK) and foetal calf serum (FCS) from
Autogenbioclean (UK Ltd.). Isoton was from Beckman Coulter UK, Ltd.
(High Wycombe, UK), phosphate-buffered saline (PBS) and
trypsin/EDTA were obtained from Sigma-Aldrich (Irvine, Ayrshire,
UK), Cell titer 96® Aqueous One Solution cell
proliferation assay was from Promega (Madison, WI, USA), MitoProbe™
JC-1 assay kit for flow cytometry was from Invitrogen (Paisley, UK)
and Mitocasp™ (S-9101 Caspase 3/7) was from Bachem (San Carlos, CA,
USA). T25 and T75 tissue culture flasks were from Nunc™ (Denmark)
and tissue culture sterile 6-, 24- and 96-well plates from
Costar®, USA. Blotting reagents were purchased from
BioRad (CA, USA and München, Germany), with the exception of TBST
and the transfer buffer, which were prepared from their
components.
Breast cancer cell lines with different genetic
backgrounds regarding p53 and ER were used to investigate the
anti-cancer effects of AMPK. The breast cancer cell lines MCF-7
(p53+ and ER+), T47D (p53 mutant and
ER+) and MDA-MB-231 (p53 mutant and ER−) were
purchased from ECACC (Porton Down, UK).
Cell culture
Breast cancer cell lines were grown in a continuous
monolayer culture in T75 top filtered sterile tissue culture flasks
inside a sterile humidified incubator at 37°C with 5%
CO2 in air. Cells were then sub-cultured as required for
maintenance and to plate cells for experimental work.
Western blotting
Cell lines were sub-cultured, counted and plated at
1×106 cells in appropriate tissue culture plates and
incubated at 37°C with 5% CO2 in air. Cells were then
incubated with AICAR (0.83 mM) or phenformin (2 mM) with
corresponding controls for 1 h. Cell lysates were collected from
the control and treated cells, followed by a protein assay. Equal
protein concentrations of control-, AICAR- and phenformin-treated
samples were loaded at a maximum volume of 30 μl/well into
pre-formed tris-HCL-SDS gels (BioRad). Proteins were then separated
by electrophoresis and transferred to a PVDF membrane. The membrane
was then blocked overnight in 10% milk prior to being incubated
overnight at 4°C with anti-phospho AMPK antibody (Cell Signaling
Technology, Danvers, MA, USA) directed against the crucial Thr172
amino acid residue. The membrane was washed in TBST four times for
15 min each and incubated with polyclonal anti-rabbit IgG antibody
(Cell Signaling Technology, #7074) and then washed again four times
prior to the addition of the fluorescence substrate. Band density
was then estimated using imaging system tools (Syngene, G: Box,
Geneflow, UK).
Cell proliferation
Cells were sub-cultured, counted by the Coulter cell
counter (Coulter Electronics ZI, Luton, Bedfordshire, UK), plated
at 7×103 cells/well in sterile 96-well plates and
incubated overnight at 37°C with 5% CO2 in air. The
following day, serial concentrations of AICAR (0–1.66 mM) or
phenformin (0–4 mM) were added. The number of viable cells was then
estimated following 72 h of incubation via their metabolic activity
using the Aqueous One assay kit (Promega), which assesses the
ability of cells to reduce the colourless tetrazolium, producing
red-coloured formazan, whose colour was measured at 490 and 630 nm
using a Biotek ELX800 Microplate reader (Biotek Instruments,
Winooski, VT, USA).
Microscopy
The cell lines were treated with 0.83 mM AICAR or 2
mM phenformin after being sub-cultured, counted and plated at
2×105 cells/well in sterile tissue culture 6-well
plates. Treated cells and corresponding controls were then examined
for morphological features of apoptosis following incubation
periods of 24, 48 and 72 h under the Xli-Cap software linked light
microscope (Xli, CETi, UK). Images were collected after 24 h for
control-, AICAR- and phenformin-treated cells, followed by
re-incubation in the same culture conditions, the process being
repeated after 48 and 72 h of incubation.
Effect of AMPK activation on
mitochondrial membrane potential
Mitochondrial membrane potential was assessed using
a fluorescence activated cell sorter (FACSCalibur, BD Biosciences,
NJ, USA). The principle involves the use of a dye (JC-1) that
accumulates inside mitochondria with intact membrane potential,
producing a red fluorescence. Depolarisation results in leakage of
the dye with a concomitant decrease in red fluorescence.
Breast cancer cell lines were sub-cultured, counted
and plated at 2×105 cells/well in sterile 6-well tissue
culture plates and incubated overnight at 37°C with 5%
CO2. The medium was removed and the cells were then
washed and treated with AICAR (0.83 mM) or phenformin (2 mM) and
incubated with corresponding controls in the same culture
conditions for 24 h. The cell monolayer was then washed with PBS
(retaining the detached cells) followed by the release of the
monolayer by trypsin/EDTA. The detached cells were then combined
with their corresponding released cells and pelleted by
centrifugation at 200 × g at 4°C for 10 min. Cell pellets were then
washed twice in the provided washing buffer by being subjected to
centrifugation at 200 × g at 4°C for 10 min. The cells were then
re-suspended in 300 μl washing buffer prior to the addition of 10
μl of fluorochrome. Labelled cells were then incubated at 37°C with
5% CO2 for 30–60 min until use for flow cytometric
analysis with appropriate filters.
Results
Western blotting
To confirm that AICAR and phenformin activated AMPK
in these cell lines, the effect of the two agents on the
phosphorylation of the enzyme by Western blotting was investigated.
The immunoblotting results revealed that AMPK in treated cells was
more phosphorylated than untreated control cells (Fig. 1A–C). No discernable difference was
evident in the total AMPK between each of the cell lines (data not
shown).
Cell proliferation
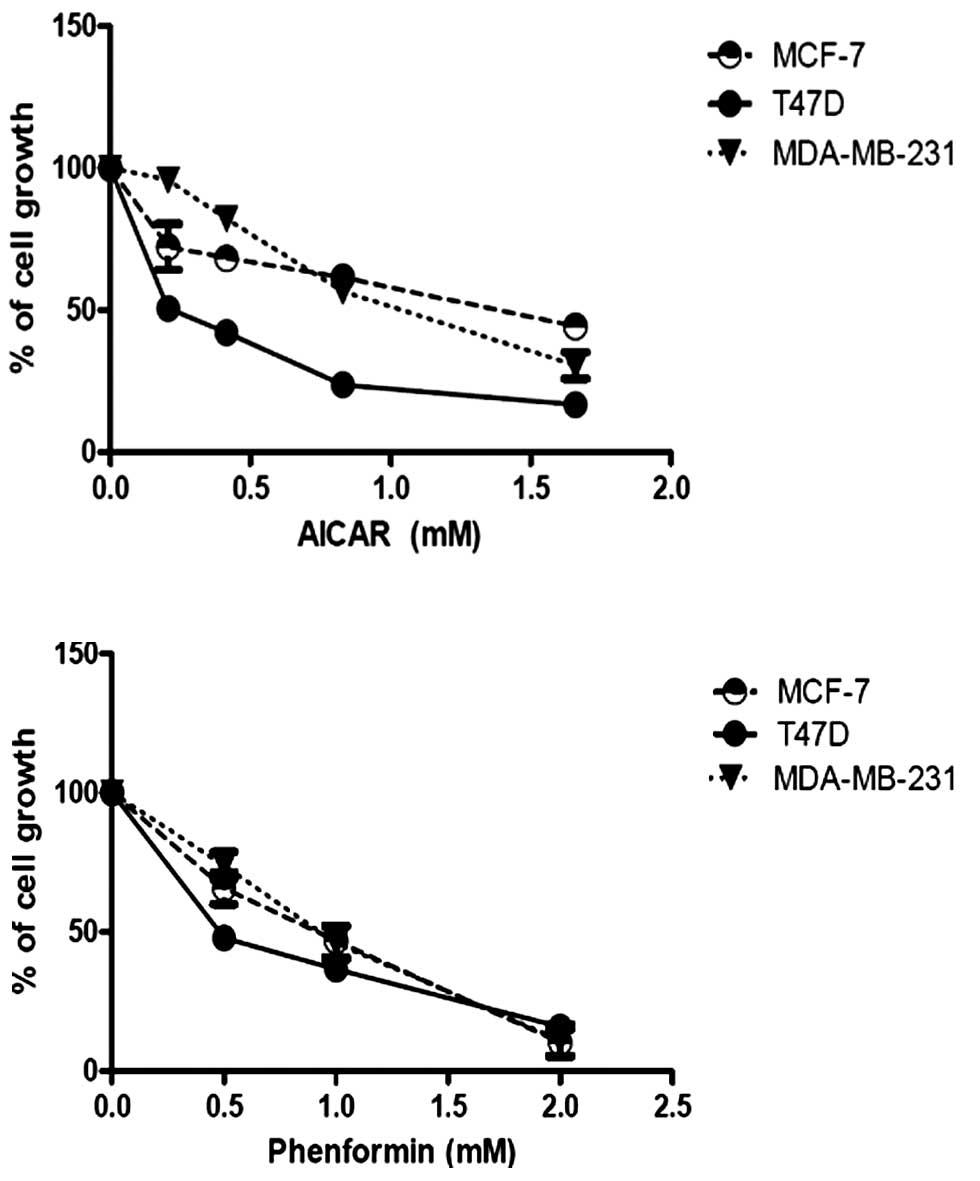
Increasing doses of the AMPK activator (AICAR)
resulted in dose responsive decreases in the proliferation of all 3
breast cancer cell lines (Fig. 2A).
T47D cells appeared to be the most sensitive to the action of
AICAR, whereas the MDA-MB-231 cells were least sensitive.
Phenformin also inhibited proliferation of the breast cancer cells,
the effects being similar in all three cell types (Fig. 2B). The maximum effect of phenformin
was greater than that of AICAR, which may be attributed, at least
in part, to its high lipid/water partition coefficient over
AICAR.
Microscopy
Over time, AMPK activation by AICAR or phenformin
induced typical apoptotic morphological features including membrane
blebbing and eventually fragmentation of cells into apoptotic
bodies in MCF-7, T47D and MDA-MB-231 cells (data not shown). It was
clear that the effect of AICAR was much slower than that of
phenformin. This slower effect of AICAR may be due to the different
pharmacokinetic properties between AICAR and phenformin, as it is
less lipophilic. Of note, the apoptotic response to AMPK activation
was lower in T47D cells than in the MCF-7 and MDA-MB-231 cell
lines, with T47D cells having the highest FasL expression.
Mitochondrial membrane potential
The activation of AMPK was found to induce
mitochondrial membrane depolarisation in all three cell types
(Fig. 3). While phenformin resulted
in almost complete depolarisation, the effect of AICAR was
sub-maximal. Notably, AMPK activation by AICAR and phenformin
disrupted mitochondrial membrane potential with a significant
difference between control- and AICAR- or phenformin-treated cells;
MCF-7 (control AICAR, p=0.016 and control phenformin, p=0.0001),
T47D (control AICAR, p=0.015 and control phenformin, p<0.0001)
and MDA-MB-231 (control AICAR, p<0.0001 and control phenformin,
p<0.0001). In addition, we have confirmed the finding of Yang
et al (12) that basal
caspase 3 and/or 7 activity has been detected in breast cancer cell
lines in the absence of any apoptotic stimuli, as indicated by the
presence of activated caspases 3 and 7 in the control as well as
AICAR- and phenformin-treated cells.
Discussion
It is now clear that AMPK is not just a family of
kinases involved in metabolic regulation by switching on and off
metabolic pathways to control the energy status inside cells.
Although not enough is known about the complete roles of AMPK, it
is realised that AMPK is a multifunctional regulatory protein whose
functions involve central (13) and
peripheral regulation. It was shown that activation of AMPK steered
three breast cancer cell lines with differing genetic backgrounds
towards cell death and functioned as an anti-proliferation agent.
This anti-proliferative effect of AMPK activation has been reported
to be via its ability to activate p53 (7) and p21 (8), thereby interfering with the cell
cycle. Although numerous studies have involved p53 in the
pro-apoptotic action of AMPK, we report a similar effect in a
breast cancer cell line, MDA-MB-231, that lacks wild-type p53. This
suggests a p53-independent pro-apoptotic action. In the present
study, T47D cells (also lacking wild-type p53) were the most
sensitive to the effect of AICAR. However, in T47D cells, the
effect appeared to be via cell cycle arrest more than via
apoptosis.
In other studies, AICAR had a cytotoxic effect on
acute lymphoblastic leukemia cells, an action that was correlated
with AMPK activation (14). A
notable finding was that AMPK activation had an anti-apoptotic
effect in normal brain cells, whereas, it was pro-apoptotic in
tumour brain cells (15).
Three main apoptotic pathways have been identified
thus far: extrinsic; intrinsic or mitochondrial; and the T
cell-mediated pathways. All pathways require involvement of certain
aspartate proteases that catalyse limited proteolysis of proteins
at aspartic acid and are collectively known as caspases. Caspases
are either initiators (caspase 2, 8, 9 and 10) or executioners
(also called effectors; caspase 3, 6 and 7). The mitochondrial or
intrinsic pathway requires the release of cytochrome C from the
mitochondria, an event that requires a prior disruption of the
mitochondrial membrane potential. This event is followed by
activation of initiator caspases that ultimately activate the
executioner caspases 3 and 7 in a cascade of limited proteolysis
(16).
In the present study, the mitochondrial or intrinsic
apoptotic pathway was found to be another target for AMPK. We
observed that in the breast cancer cell lines (with and without p53
or ER), AMPK activation resulted in disruption of the mitochondrial
membrane potential. These results indicate that activation of AMPK
potentially induces apoptosis via a p53 or p21-independent pathway.
It has been reported that breast cancer cell lines have high basal
levels of caspase activation, which aid in cell migration and
metastasis, completely independently of any apoptotic stimuli
(12).
Mitochondrial respiratory complex 1 has been
reported to be a target of metformin and thialozidinediones (TZDs)
that are widely used to treat diabetic patients suffering from
severe insulin resistance. Inhibition by metformin of the
mitochondrial respiratory complex 1 and the resultant impairment of
mitochondrial function and cellular respiration has been thought to
be responsible for its anti-diabetic effect. The mechanism of
action of metformin was not clear until the report (17) that showed that AMPK mediates
metformin action, and that inhibition of AMPK eliminated the effect
of metformin. This mechanism indicates that the mitochondrial
membrane potential may be affected by activated AMPK and trigger
the intrinsic pathway of apoptosis by enhancing the release of the
pro-apoptotic factors from mitochondria. This action is supported
by our study on three breast cancer cell lines differing in their
genetics regarding p53 and ER status.
The enhanced effect of phenformin as a mitochondrial
membrane potential disruptor may be attributed to the previously
reported affinity of biguanides, including phenformin, to
accumulate inside mitochondria, an action that has not been
reported for AICAR. These observations suggest that the effect of
AICAR on the mitochondrial membrane potential may be mediated by
AMPK. However, the effect is slower than that of phenformin due to
differences in their pharmacokinetic properties. The effect of
phenformin could be direct or indirect via an ATP
decrease-dependent activation of AMPK.
MDA-MB-231 cells were the least sensitive in most
assessments; however, AMPK activation in MDA-MB-231 cells induced
mitochondrial membrane potential disruption to the same degree as
that noted in other cell lines. It is unlikely that the p53 status
of the MDA-MB-231 cells is responsible for their low sensitivity to
AMPK-induced growth inhibition, since the most sensitive cells,
T47D, also express mutant p53. However, further investigations are
required to delineate the underlying reasons. In addition, T47D
cells were the least sensitive to apoptotic changes, as was
observed in photomicrographs. Unlike MCF-7 and MDA-MB-231 cells,
the cell cycle analysis of T47D cells revealed a cell cycle arrest
effect of AMPK activation by AICAR and phenformin rather than the
apoptotic effect which was more apparent in the MCF-7 and
MDA-MB-231 cell lines.
The greater sensitivity of T47D cells may be related
to the fact that Western blotting revealed two bands reacting to
the phospho-AMPK antibody, whereas only one band was observed in
lysates from MCF-7 and MDA-MB-231 cells.
In conclusion, we have observed significantly
different effects of AMPK activation on the growth and death of
three human breast cancer cell lines. Thus far, it is not possible
to ascribe these differences to p53 status or ER expression, which
are key differences that have been reported previously. However,
further studies are required to ascertain the intracellular
signalling pathways responsible, and the possible interactions
(crosstalk) with other signals. Furthermore, the extent to which
AMPK may be a target or co-target for therapeutic intervention in
breast cancer should be explored.
References
|
1
|
Evans DG and Howell A: Breast cancer
risk-assessment models. Breast Cancer Res. 9:2132007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lorincz AM and Sukumar S: Molecular links
between obesity and breast cancer. Endocr Relat Cancer. 13:279–292.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Todorov PT, McDevitt TM, Meyer DJ, Ueyama
H, Ohkubo I and Tisdale MJ: Purification and characterization of a
tumor lipid-mobilizing factor. Cancer Res. 58:2353–2358.
1998.PubMed/NCBI
|
|
4
|
Katajisto P, Vallenius T, Vaahtomeri K,
Ekman N, Udd L, Tiainen M and Makela TP: The LKB1 tumor suppressor
kinase in human disease. Biochim Biophys Acta. 1775:63–75.
2007.PubMed/NCBI
|
|
5
|
Lizcano JM, Goransson O, Toth R, Deak M,
Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG and
Alessi DR: LKB1 is a master kinase that activates 13 kinases of the
AMPK subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Viollet B, Athea Y, Mounier R, Guigas B,
Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J,
Beauloye C, Foretz M, Andreelli F, Ventura-Clapier R and Bertrand
L: AMPK: Lessons from transgenic and knockout animals. Front
Biosci. 14:19–44. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bode AM and Dong Z: Post-translational
modification of p53 in tumorigenesis. Nat Rev Cancer. 4:793–805.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Caraci F, Chisari M, Frasca G, Chiechio S,
Salomone S, Pinto A, Sortino MA and Bianchi A: Effects of
phenformin on the proliferation of human tumor cell lines. Life
Sci. 74:643–650. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo Z, Saha AK, Xiang X and Ruderman NB:
AMPK, the metabolic syndrome and cancer. Trends Pharmacol Sci.
26:69–76. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiang X, Saha AK, Wen R, Ruderman NB and
Luo Z: AMP-activated protein kinase activators can inhibit the
growth of prostate cancer cells by multiple mechanisms. Biochem
Biophys Res Commun. 321:161–167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rattan R, Giri S, Singh AK and Singh I:
5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits
cancer cell proliferation in vitro and in vivo via AMP-activated
protein kinase. J Biol Chem. 280:39582–39593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang L, Cao Z, Yan H and Wood WC:
Coexistence of high levels of apoptotic signaling and inhibitor of
apoptosis proteins in human tumor cells: implication for cancer
specific therapy. Cancer Res. 63:6815–6824. 2003.PubMed/NCBI
|
|
13
|
Butler H and Korbonits M: Cannabinoids for
clinicians: the rise and fall of the cannabinoid antagonists. Eur J
Endocrinol. 161:655–662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sengupta TK, Leclerc GM, Hsieh-Kinser TT,
Leclerc GJ, Singh I and Barredo JC: Cytotoxic effect of
5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on
childhood acute lymphoblastic leukemia (ALL) cells: implication for
targeted therapy. Mol Cancer. 6:462007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mukherjee P, Mulrooney TJ, Marsh J, Blair
D, Chiles TC and Seyfried TN: Differential effects of energy stress
on AMPK phosphorylation and apoptosis in experimental brain tumor
and normal brain. Mol Cancer. 7:372008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N,
Hirshman MF, Goodyear LJ and Moller DE: Role of AMP-activated
protein kinase in mechanism of metformin action. J Clin Invest.
108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|