Introduction
Gastrointestinal stromal tumours (GISTs) are the
most common mesenchymal neoplasms of the gastrointestinal tract and
are believed to originate from the interstitial cells of Cajal
(1–3). GISTs are characterised by the
expression of the type III receptor tyrosine kinase KIT encoded by
the KIT proto- oncogene (4),
thus KIT immunohistochemistry has been used as a diagnostic marker
of GISTs (4,5). Since Hirota et al (6) first reported the KIT mutation
in 1998, a number of studies have reported that the majority of
GISTs have oncogenic mutations in KIT (7,8). A
small subset of GISTs was also found to possess activating
mutations in the platelet-derived growth factor receptor A
(PDGFRA) gene (9,10).
Therapeutic targeting with the tyrosine kinase
inhibitor imatinib (Glivec, STI571) has been shown to be effective
in patients with advanced or unresectable GISTs (11,12).
Imatinib is an inhibitor of a number of tyrosine kinases, including
the intracellular kinase ABL, the growth factor receptors KIT and
PDGFR-A and -B and their oncogenic activated forms (13). Imatinib competes with adenosine
triphosphate (ATP) for the ATP-binding site of the kinases,
preventing downstream signalling (14,15).
Imatinib is now successfully used in the treatment of advanced
GISTs (16,17) and chronic myelogenous leukaemia
(18,19). Its clinical activity in other
neoplasms has also been reported, including chordoma (20) and dermatofibrosarcoma protuberans
(21).
Evidence suggests that different types of mutation
in GISTs correlate with different response rates to imatinib
(22). Previous studies have
demonstrated that the majority of GISTs with a KIT mutation
were sensitive to imatinib, but GISTs with a PDGFRA mutation
were mostly resistant to imatinib. Most of the PDGFRA
mutations found in GISTs have been identified in exon 18 and are
the imatinib-resistant substitution D842V. Only approximately
one-third of PDGFRA mutations in GISTs have been found in
exons 12 and 14 and these mutations have different responses to
imatinib (23). We previously
studied the PDGFRA mutation types of exon 12 and 18 by PCR
amplification and DNA sequencing in a series of Chinese GISTs.
Compared with the reports in the published data, a point mutation
at codon 839, L839P, lies outside the hot spot area (23). Further studies confirmed that
PDGFRAL839P is a gain-of-function mutation
(24). To explore the response of
PDGFRAL839P to imatinib, we transfected different
isoforms of the human PDGFRA gene into Chinese hamster ovary
(CHO) cells and compared the inhibitory effects of imatinib on
PDGFRAL839P with the effects on
PDGFRAD842V in vitro concerning cell
growth, apoptosis and receptor phosphorylation level.
Materials and methods
PDGFRA expression constructs
The wild-type human PDGFRA cDNA (HD
Biosciences Co, Shanghai, China) was cloned into the pcDNA3.1hygro+
vector to create pcDNA3.1-PDGFRAWild. cDNA encoding the
human PDGFRA mutant isoforms was generated using a MutanBEST
site-specific mutagenesis kit (Takara Bio, Inc., Shiga, Japan)
using the primers: 5′-CTGTGACTTTGGCCCGGCCAGAGACATCATG-3′ and
5′-CATGATGTCTCTGGCCGGGCCAAAGTCACAG-3′ for the
PDGFRAL839P cDNA, and 5′-GGCCTGGCCAGAGT
CATCATGCATGATTCG-3′ and 5′-CGAATCATGCATGAT GACTCTGGCCAGGCC-3′ for
the PDGFRAD842V cDNA. A ~3.3-kb product was
obtained by denaturation for 1 min at 94°C, annealing for 1 min at
56°C and extension for 1 min at 72°C for 30 cycles. These fragments
were digested with XhoI and NheI and directionally
cloned into the pcDNA3.1hygro+ vector. All vectors were confirmed
by restriction endonuclease digestion and bidirectional
sequencing.
Transfection of CHO cell lines
To transfect CHO cells with plasmids encoding human
PDGFRAWild, PDGFRAD842V and
PDGFRAL839P, LipofectamineTM 2000
(Invitrogen, Carlsbad, CA, USA) was used according to the
manufacturer's instructions. Infectants were selected with 1 mg/ml
Hygromycin B (Merck, Darmstadt, Germany) until all the uninfected
control cells were killed. Following selection, the CHO cells were
cultured in medium containing 0.5 mg/ml Hygromycin B.
Flow cytometric analysis of
apoptosis
The CHO cell lines were cultured in the presence or
absence of imatinib for 24 h. Subsequently, apoptosis was detected
using an annexin V-fluorescein isothiocyanate (FITC) staining kit
(Roche, Mannheim, Germany). The cells were harvested by
trypsinisation and labelled with annexin V-FITC for 15 min at 4°C
and analysed by FACSCalibur flow cytometry.
Methyl thiazolyl tetrazolium (MTT)
assay
Cells were added to 96-well plates at a density of
2×104 cells/well. After the cells were maintained for 24
h at 37°C in a 5% CO2 atmosphere, a 200 μl solution
containing imatinib (0, 0.001, 0.01, 0.1, 1 or 5 μM) was added.
After 72 h, 20 μl of a 5 mg/ml solution in PBS of the MTT (Sigma,
St. Louis, MO, USA) tetrazolium substrate was added and the cells
were incubated for 4 h at 37°C. The resulting violet formazan
precipitate was solubilised by the addition of 150 μl DMSO
(Amresco, Solon, OH, USA) and incubated for 10 min at room
temperature. Sample absorbances were then measured on a plate
reader at 540 nm.
Western blotting
Whole-cell lysates were prepared by resuspending the
cells in cold SDS buffer [1% SDS, 0.04 mol/l Tris-HCl (pH 6.8), 5%
glycerol]. The protein concentrations were determined using a
commercial BCA protein assay kit (Merck). Protein extracts were
added at a 4:1 ratio to 5X SDS sample buffer and boiled. The
protein (50 μg) was resolved on a 10% polyacrylamide gel and
transferred to polyvinylidene difluoride membranes (Millipore,
Billerica, MA, USA). The membranes were probed with anti-PDGFRA
rabbit polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), anti-GAPDH goat polyclonal antibody (GenScript,
Piscataway, NJ, USA) or anti-phosphotyrosine monoclonal antibody
(Long Island Biotech Inc., New York, USA). The signals were
detected using the western blotting luminal reagent (Santa Cruz
Biotechnology, Inc.).
Results
Identification of recombinant
plasmids
Following digestion by XhoI and NheI,
bands at 3.3 kDa were detected for the positive clones, suggesting
that PDGFRAWild, PDGFRAL839P
and PDGFRAD842V fragments were inserted into the
pcDNA3.1 vector, designated as recombinant plasmids
pcDNA3.1-PDGFRAWild, pcDNA3.1-PDGFRAL839P and
pcDNA3.1-PDGFRAD842V, respectively.
pcDNA3.1-PDGFRAWild,
pcDNA3.1-PDGFRAL839P and pcDNA3.1-PDGFRAD842V
DNA was prepared for sequencing. The sequence obtained was the same
as the reported sequence of PDGFRA cDNA and mutant
PDGFRA cDNA, indicating that the wild-type and mutant
PDGFRA genes were successfully cloned into the eukaryotic
expression vector pcDNA3.1.
Liposome transfer of
PDGFRAWild, PDGFRAD842V and
PDGFRAL839P into CHO cell lines
CHO cells were transfected with
LipofectamineTM 2000 encoding human
PDGFRAWild, PDGFRAD842V and
PDGFRAL839P. Following selection in medium
containing 1 mg/ml Hygromycin B, the CHO cells were raised in
medium containing 0.5 mg/ml Hygromycin B. The expression of human
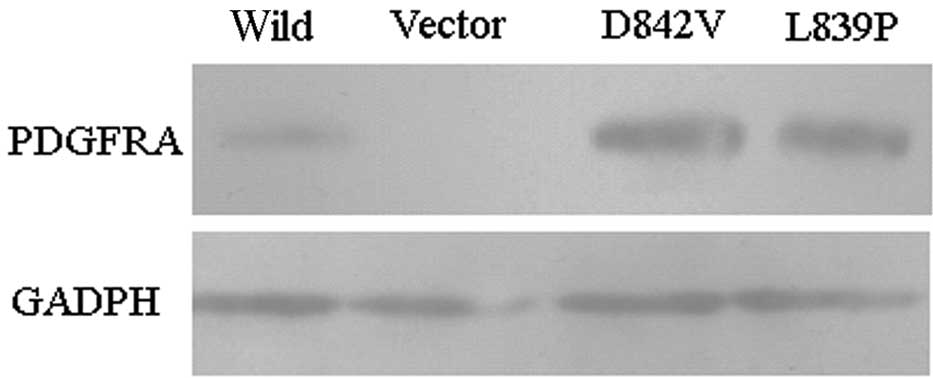
PDGFRA was examined using western blot analysis (Fig. 1). Results indicated that human
PDGFRA was expressed in CHO(Wild), CHO(D842V) and CHO(L839P) cells,
but not CHO(vector) cells. The level of expression of human PDGFRA
protein in the CHO(D842V) and CHO(L839P) cells was higher than that
in the CHO(Wild) cells.
Mutation isoforms of the PDGFRA gene
affect the sensitivity to imatinib
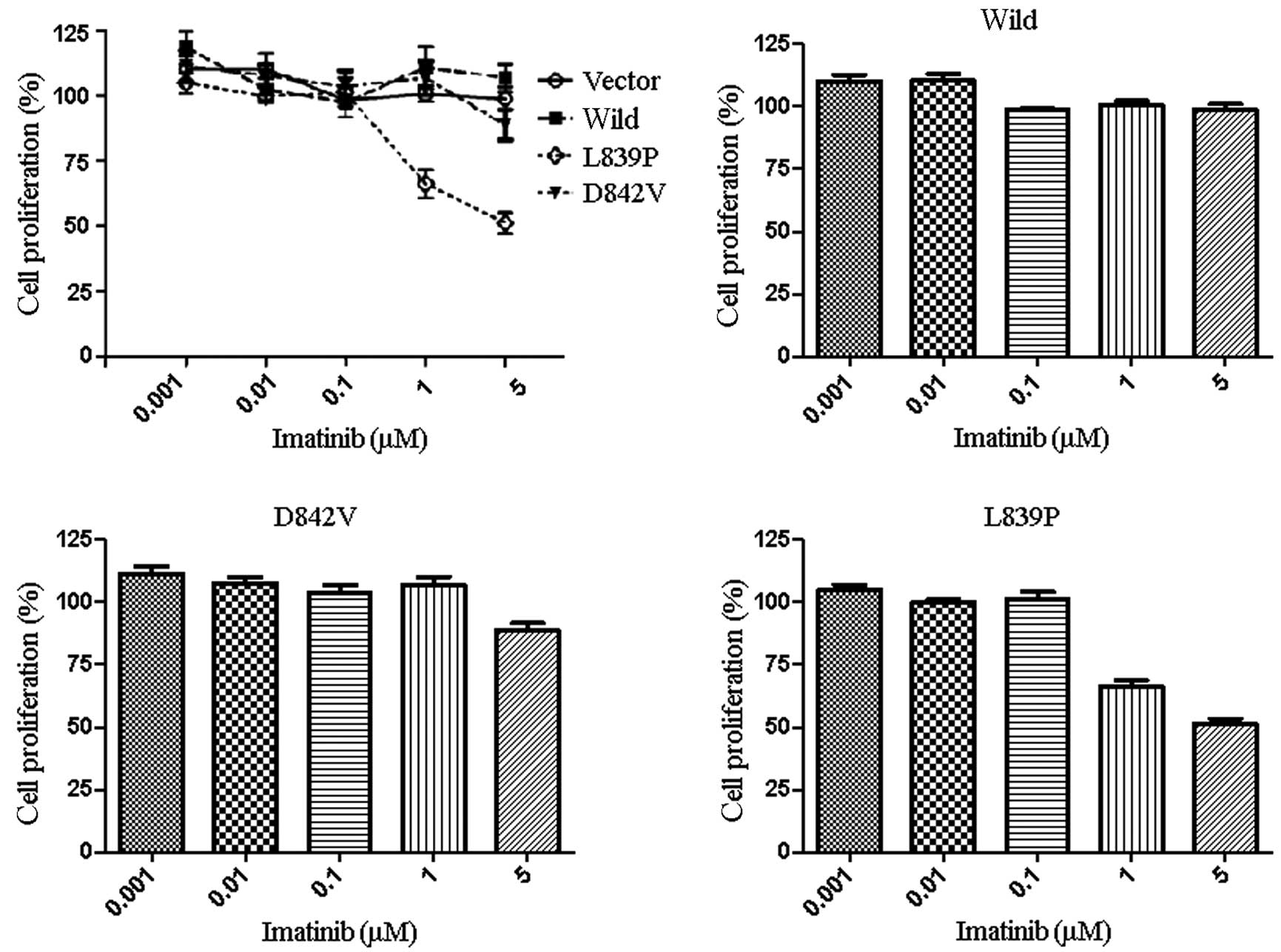
The effect of imatinib on the growth of CHO(vector),
CHO(Wild), CHO(D842V) and CHO(L839P) cells was evaluated using an
MTT assay that measures the number of live cells at the end of a
72-h culture period. The proliferation of CHO(L839P) cells was
inhibited by imatinib at concentrations of 1 μM (Fig. 2A and D). By contrast, the remaining
cell lines, including CHO(D842V), were not greatly affected by the
presence of imatinib at concentrations of ≥5 μM (Fig. 2A-C). These results suggest that the
CHO(L839P) cell line is more sensitive to inhibition by imatinib
than CHO(D842V) and CHO(Wild) cells, indicating that the L839P
mutation of the PDGFRA gene may be sensitive to imatinib,
but the D842V mutation is resistant to imatinib.
Imatinib induces apoptosis in CHO
cells
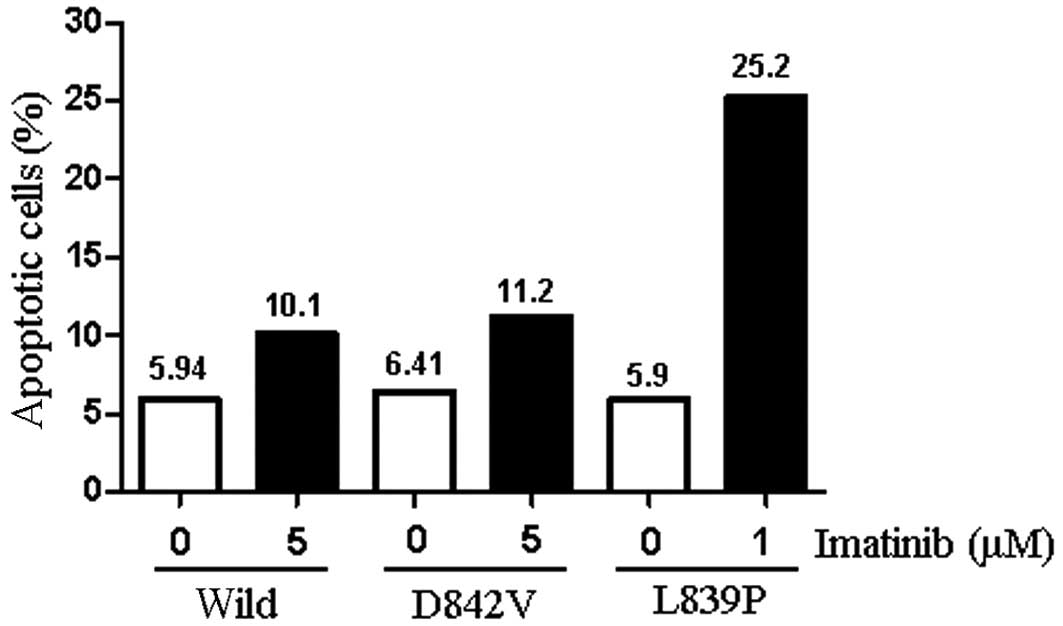
To determine whether growth inhibition was caused by
the induction of apoptosis, staining with Annexin-V and propidium
iodide was used. Following the incubation of CHO(vector) cells in 5
μM imatinib, the percentage of apoptotic cells remained ~10% of the
total population. By contrast, incubation of CHO(L839P) in 1 μM
imatinib resulted in a ~4-fold increase in the percentage of
apoptotic cells. Although the incubation of CHO(D842V) and
CHO(Wild) cells in 5 μM imatinib resulted in a slight increase in
the number of apoptotic cells, the percentage of apoptotic cells
remained ~10% of the total population (Fig. 3). Similar results were obtained when
the experiment was repeated.
Effect of imatinib on receptor
phosphorylation
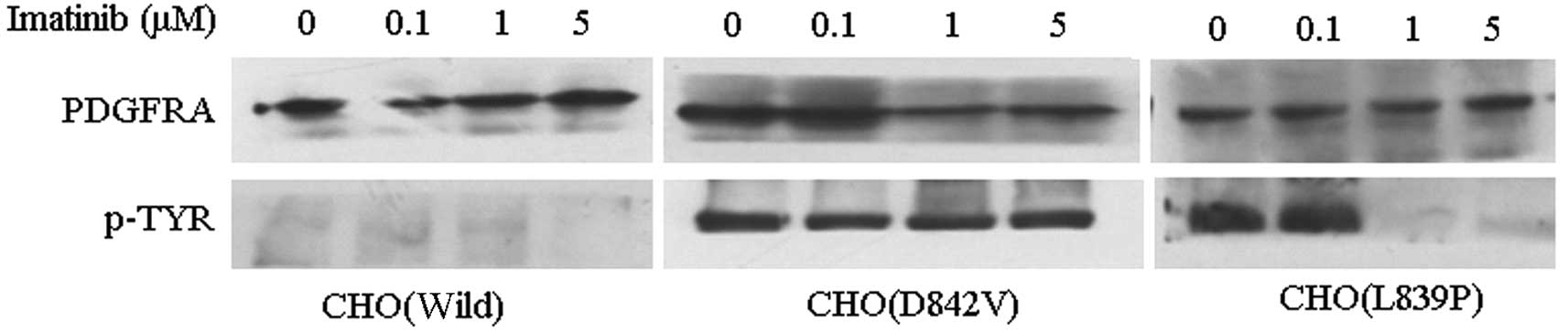
To determine the effect of imatinib on the
autophosphorylation of PDGFRA, a western blot for PDGFRA and
phosphotyrosine was performed (Fig.
4). The phosphotyrosine levels revealed that PDGFRA
phosphorylation in CHO(Wild) cells was low. By contrast, the
receptor phosphorylation in CHO(D842V) was unaffected by 5 μM
imatinib. There was no phosphorylation of PDGFRA in CHO(L839P)
cells treated with 1 and 5 μM imatinib. These results correlate
well with those of the MTT assays, indicating that CHO(L839P) cells
are sensitive to 1 μM imatinib, whereas CHO(D842V) cells are
resistant to imatinib.
Discussion
GISTs are the most common mesenchymal neoplasms of
the gastrointestinal tract (1–3).
Despite clinicopathological differences, most GISTs share a similar
genetic profile, including KIT or PDGFRA gene
gain-of-function mutations (25,26),
which are targets for the kinase inhibitor imatinib (13,27).
Previous clinical studies have shown that imatinib is effective in
patients with advanced or unresectable GISTs (11,12).
Further studies have demonstrated that different responses of GIST
patients to imatinib correlate with different mutation types
(22,28). Results of the study by Corless et
al suggest that approximately 62.6% of PDGFRA-mutant tumours
are resistant to imatinib (23). In
other words, only just over a third of GISTs with PDGFRA mutations
may respond to imatinib. Therefore, mutation screening may aid in
the management of GIST patients.
In our previous studies, a novel point mutation in
exon 18 of the PDGFRA gene (L839P) was found in two GIST
cases (24) and lies outside of the
hot spot. To compare the inhibitory effects of imatinib on
PDGFRAL839P with the effects on
PDGFRAD842V in vitro with the mutations
expressed individually in the same cellular background, we used
liposome transduction to transfect these forms of PDGFRA
into CHO cells. The D842V mutation is the most common mutation of
the PDGFRA gene in GISTs. Preliminary data suggested that
D842V is resistant to imatinib in vitro and in vivo
(22). In the present study, the
MTT assay results indicated that the PDGFRA mutant isoform
D842V shows significant resistance to imatinib at 1 μM, which is
equivalent to the highest serum levels generally achieved in
patients, according to other studies (29). Compared with D842V, the L839P
mutation of the PDGFRA gene was sensitive to 1 μM imatinib
in vitro, suggesting that GISTs carrying the
PDGFRAL839P mutation may have a better response
to imatinib. To determine whether growth inhibition was caused by
the induction of apoptosis, staining with Annexin-V was used. The
results suggest that imatinib notably induces cell apoptosis in
CHO(L839P) cells and enhances the response to imatinib.
To determine the effect of imatinib on
autophosphorylation of PDGFRA, western blots for PDGFRA and
phosphotyrosine were performed. The results indicate that the
differences in the sensitivity of PDGFRA mutants to imatinib
are a direct consequence of the ability or inability of imatinib to
inhibit PDGFRA phosphorylation.
In conclusion, to compare the inhibitory effects of
imatinib on PDGFRAD842V and
PDGFRAL839P with the effects on
PDGFRAWild, with the mutations expressed
individually in the same cellular background, we used liposome
transduction to transfect these forms of PDGFRA into CHO
cells. Our data concerning cell growth, apoptosis and receptor
phosphorylation indicate that the D842V mutant was resistant to
imatinib, which is consistent with the results of previous studies
(22,23). Our data also demonstrate that a new
mutant, PDGFRAL839P, was more sensitive to
imatinib than PDGFRAWild and
PDGFRAD842V. This study suggests that screening
patients for PDGFRA mutations is essential to identify malignancies
that are likely to be sensitive or resistant to treatment with
imatinib.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (project numbers 30700809 and
30972876).
Abbreviations:
|
ATP
|
adenosine triphosphate
|
|
FITC
|
fluorescein iso- thiocyanate
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
GIST
|
gastrointestinal stromal tumour
|
|
MTT
|
methyl thiazolyl tetrazolium
|
|
PBS
|
phosphate-buffered solution
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
References
|
1
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: review on morphology, molecular pathology,
prognosis, and differential diagnosis. Arch Pathol Lab Med.
130:1466–1478. 2006.PubMed/NCBI
|
|
2
|
Meng L, Fang SH and Jin M: An unusual case
of pancreatic and gastric neoplasms (2010: 12b). Malignant GISTs
originating from the pancreas and stomach. Eur Radiol. 21:663–665.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huizinga JD, Thuneberg L, Klüppel M,
Malysz J, Mikkelsen HB and Bernstein A: W/kit gene required for
interstitial cells of Cajal and for intestinal pacemaker activity.
Nature. 373:347–349. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarlomo-Rikala M, Kovatich AJ,
Barusevicius A and Miettinen M: CD117: a sensitive marker for
gastrointestinal stromal tumors that is more specific than CD34.
Mod Pathol. 11:728–734. 1998.PubMed/NCBI
|
|
5
|
Fletcher CD, Berman JJ, Corless C,
Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti
H, Rubin BP, et al: Diagnosis of gastrointestinal stromal tumors: A
consensus approach. Hum Pathol. 33:459–465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rubin BP, Singer S, Tsao C, Duensing A,
Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, et al: KIT
activation is a ubiquitous feature of gastrointestinal stromal
tumors. Cancer Res. 61:8118–8121. 2001.PubMed/NCBI
|
|
8
|
Corless CL, Fletcher JA and Heinrich MC:
Biology of gastrointestinal stromal tumors. J Clin Oncol.
22:3813–3825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hirota S, Ohashi A, Nishida T, Isozaki K,
Kinoshita K, Shinomura Y and Kitamura Y: Gain-of-function mutations
of platelet-derived growth factor receptor alpha gene in
gastrointestinal stromal tumors. Gastroenterology. 125:660–667.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Demetri GD, von Mehren M, Blanke CD, Van
den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA,
Singer S, Janicek M, et al: Efficacy and safety of imatinib
mesylate in advanced gastrointestinal stromal tumors. N Engl J Med.
347:472–480. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Oosterom AT, Judson I, Verweij J,
Stroobants S, Donato di Paola E, Dimitrijevic S, Martens M, Webb A,
Sciot R, Van Glabbeke M, Silberman S and Nielsen OS; European
Organisation for Research and Treatment of Cancer Soft Tissue and
Bone Sarcoma Group. Safety and efficacy of imatinib (STI571) in
metastatic gastrointestinal stromal tumours: a phase I study.
Lancet. 358:1421–1423. 2001.PubMed/NCBI
|
|
13
|
Tamborini E, Bonadiman L, Greco A,
Albertini V, Negri T, Gronchi A, Bertulli R, Colecchia M, Casali
PG, Pierotti MA and Pilotti S: A new mutation in the KIT ATP pocket
causes acquired resistance to imatinib in a gastrointestinal
stromal tumor patient. Gastroenterology. 127:294–299. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Buchdunger E, Cioffi CL, Law N, Stover D,
Ohno-Jones S, Druker BJ and Lydon NB: Abl protein-tyrosine kinase
inhibitor STI571 inhibits in vitro signal transduction mediated by
c-kit and platelet-derived growth factor receptors. J Pharmacol Exp
Ther. 295:139–145. 2002.
|
|
15
|
Waller CF: Imatinib mesylate. Recent
Results Cancer Res. 184:3–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cassier PA and Blay JY: Imatinib mesylate
for the treatment of gastrointestinal stromal tumor. Expert Rev
Anticancer Ther. 10:623–634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lopes LF and Bacchi CE: Imatinib treatment
for gastrointestinal stromal tumour (GIST). J Cell Mol Med.
14:42–50. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marin D: Current status of imatinib as
frontline therapy for chronic myeloid leukemia. Semin Hematol.
47:312–318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mauro MJ and Druker BJ: STI571: targeting
BCR-ABL as therapy for CML. Oncologist. 6:233–238. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Casali PG, Messina A, Stacchiotti S,
Tamborini E, Crippa F, Gronchi A, Orlandi R, Ripamonti C, Spreafico
C, Bertieri R, et al: Imatinib mesylate in chordoma. Cancer.
101:2086–2097. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kérob D, Porcher R, Vérola O, Dalle S,
Maubec E, Aubin F, D'Incan M, Bodokh I, Boulinguez S,
Madelaine-Chambrin I, et al: Imatinib mesylate as a preoperative
therapy in dermatofibrosarcoma: results of a multicenter phase II
study on 25 patients. Clin Cancer Res. 16:3288–3295.
2010.PubMed/NCBI
|
|
22
|
Heinrich MC, Corless CL, Demetri GD,
Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den
Abbeele AD, Druker BJ, et al: Kinase mutations and imatinib
response in patients with metastatic gastrointestinal stromal
tumor. J Clin Oncol. 21:4342–4349. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Corless CL, Schroeder A, Griffith D, Town
A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J and
Heinrich MC: PDGFRA mutations in gastrointestinal stromal tumors:
frequency, spectrum and in vitro sensitivity to imatinib. J Clin
Oncol. 23:5357–5364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang L, Bai CG, Hou XW, Liu XH and Ma DL:
Transforming effect of PDGFRA gene mutant on the cell function in
gastrointestinal stromal tumor. Zhonghua Zhong Liu Za Zhi.
31:500–504. 2009.(In Chinese).
|
|
25
|
Lasota J and Miettinen M: Clinical
significance of oncogenic KIT and PDGFRA mutations in
gastrointestinal stromal tumours. Histopathology. 53:245–266. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lasota J and Miettinen M: KIT and PDGFRA
mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn
Pathol. 23:91–102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frost MJ, Ferrao PT, Hughes TP and Ashman
LK: Juxtamembrane mutant V560GKit is more sensitive to Imatinib
(STI571) compared with wild-type c-kit whereas the kinase domain
mutant D816VKit is resistant. Mol Cancer Ther. 1:1115–1124.
2002.PubMed/NCBI
|
|
28
|
Merkelbach-Bruse S, Dietmaier W, Füzesi L,
Gaumann A, Haller F, Kitz J, Krohn A, Mechtersheimer G, Penzel R,
Schildhaus HU, et al: Pitfalls in mutational testing and reporting
of common KIT and PDGFRA mutations in gastrointestinal stromal
tumors. BMC Med Genet. 11:1062010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Demetri GD, Wang Y, Wehrle E, Racine A,
Nikolova Z, Blanke CD, Joensuu H and von Mehren M: Imatinib plasma
levels are correlated with clinical benefit in patients with
unresectable/metastatic gastrointestinal stromal tumors. J Clin
Oncol. 27:3141–3147. 2009. View Article : Google Scholar : PubMed/NCBI
|