Introduction
Nevoid basal cell carcinoma syndrome (NBCCS; also
known as Gorlin syndrome; OMIM no. 109400) is a rare autosomal
dominant disorder. The disease is characterized by multiple basal
cell carcinomas, keratocystic odontogenic tumors (KCOTs) of the
jaw, palmar and/or plantar pits and developmental defects and
skeletal anomalies, including bifid, fused and splayed ribs,
intracranial calcification, for example of the falx cerebri, and a
variety of other benign or malignant tumors, including ovarian
fibroma, medulloblastoma, rhabdomyosarcomas and cardiac fibromas
(1).
The incidence of NBCCS varies greatly among
countries from one case per 55,600 (2) to one case per 256,000 in the
population (3). The prevalence is
predicted to be markedly higher in individuals younger than 20
years who present with basal cell carcinomas (BCCs) (4).
Since the discovery in 1996 that NBCCS might be
associated with germline mutations in the PTCH1 gene (5,6)
approximately 280 germline mutations have been identified worldwide
(7).
The PTCH1, or human homolog 1 of the
Drosophila Patched gene, mapped on chromosome 9q22.3,
consists of 23 exons and encodes an integral membrane protein of
1,447 amino acids, Patched 1 (PTCH1) with 12 transmembrane regions,
2 extracellular loops and a putative sterol-sensing domain
(8). PTCH mutations are involved in
the formation of syndromic as well as non-syndromic keratocysts
(9). Non-syndromic KCOTs are common
benign cystic tumors of the jaw (10).
The vast majority of cysts occur sporadically and in
a single form on the upper and/or lower jaw of middle-aged
individuals. When associated with NBCCS, these lesions appear
earlier, often during the first or second decade of life, and may
be multiple (10). Loss of
heterozygosity on chromosome 9q22.3 has been described in patients
with sporadic and syndrome-associated odontogenic keratocysts
(11,12). In addition, somatic mutations in the
PTCH1 gene in odontogenic keratocysts associated with NBCCS have
previously been described (13).
Recently, a novel missense germline mutation
c.3277G>C (p.G1093R) in exon 19 of the PTCH1 gene has
been described as being responsible for non-syndromic KCOTs in a
Chinese family without evidence of NBCCS, suggesting a
genotype-phenotype correlation (14).
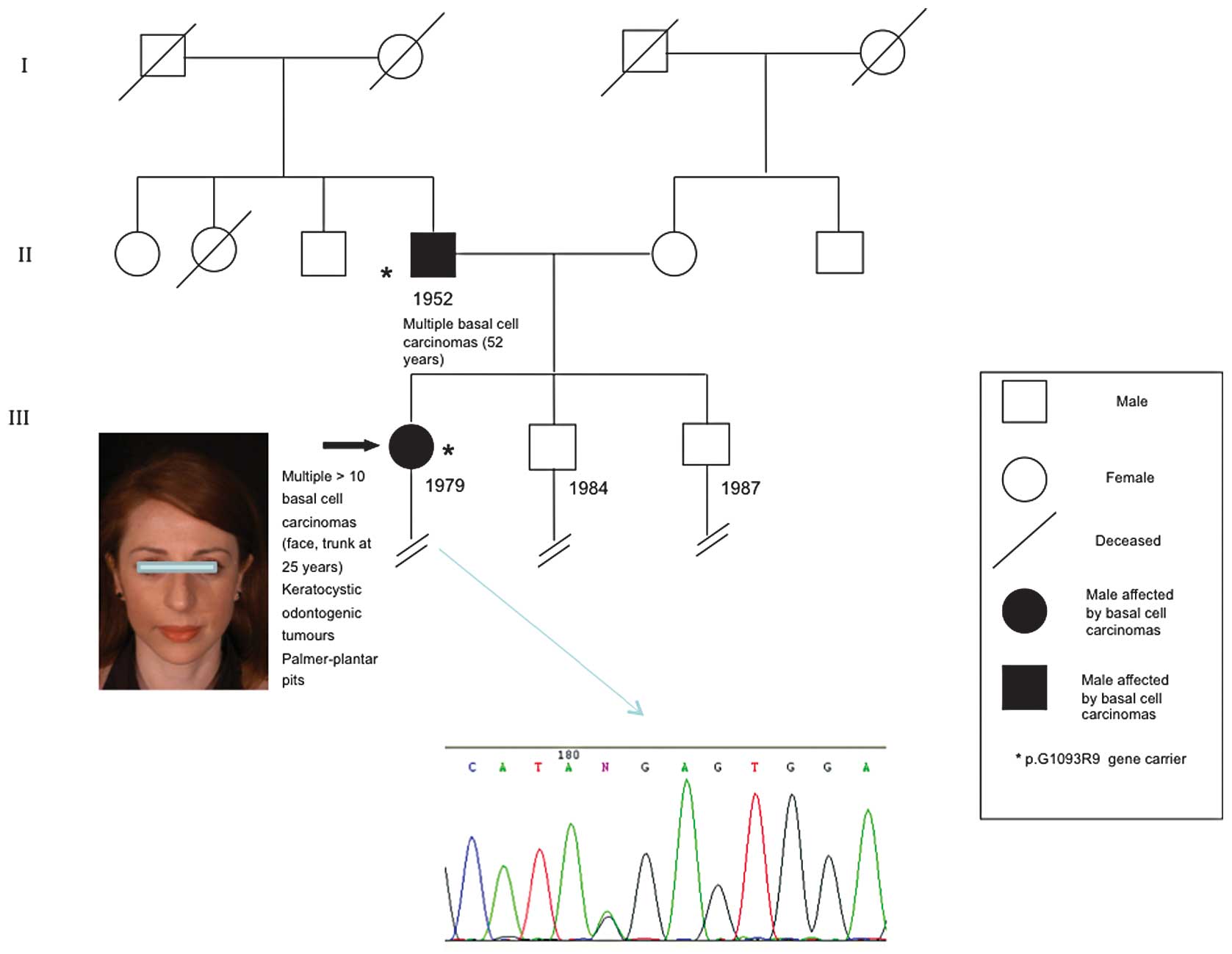
The present study reports for the first time this
same mutation (p.G1093R) in a familial case of NBCCS and KCOTs, in
which father and daughter presented with numerous basal cell
carcinomas and multiple KCOTs of the jaw. Unlike the previous study
(14), this case report widens the
phenotypic spectrum of this missense mutation.
Patients and methods
Case report
A 36-year-old woman was referred to the Department
of Dermatological and Genetic Counseling due to multiple BCCs and
KCOTs of the jaw, diagnosed at the age of 25 and 14 years,
respectively. The patient presented with low height, weight and
occipitofrontal circumference (OFC<97th percentile) within
normal limits, extremely light pigmentation of the skin and
multiple palmoplantar pits on the hands and feet. The patient also
presented with multiple papular and nodular lesions located on the
face and trunk.
On physical examination, round or oval-shaped
lesions 3–10 mm in diameter with a smooth or ulcerated and crusted
surface were observed on the face and trunk (Fig. 1). Surgical excision of numerous
lesions was performed. Histopathological examination revealed the
presence of nests of basaloid cells, arranged at the periphery of
the nodules in palisading structures. A diagnosis of multiple BCCs
was made.
The patient had undergone surgical excision of
multiple BCCs (>20) on the face and trunk over a period of 21
years of dermatological follow-up. At the age of 15, the patient
also underwent surgical enucleation of osteolytic lesions located
in her jaw and a histopathological diagnosis of keratocysts was
made. Cardiac and gynecological ultrasound evaluations did not
reveal any abnormalities, and neither did radiographical
examinations of the vertebral column and thoracic cage. Therefore,
in line with the major and minor criteria currently present in the
literature (15), a diagnosis of
NBCCS was made, as four major criteria were fulfilled, including
the presence of a first-degree relative affected by NBCCS. The
patient had been born at term from non-consanguineous parents and
had two unaffected siblings. However, the proband reported that her
52-year-old father had previously undergone surgical excision of
multiple cutaneous lesions, which had been histologically diagnosed
as BCCs. No documented evidence of KCOT was available for this
patient although the proband referred to several non-specific
dental problems. No BCCs developed among other family members.
Cytogenetic and molecular analysis
Peripheral blood samples were collected from the
proband and her father. Informed consent was obtained from the two
patients. Molecular analysis of PTCH1 was performed as previously
described (3). The PTCH1 cDNA
sequence from GenBank (Accession number U59464.1) was used as a
reference sequence, where the A of the ATG translation initiation
start site represents nucleotide +1.
Sequencing analysis of the constitutional DNA of the
proband and her father led to the identification of the same exon
19 missense mutation of PTCH1 (p.G1093R). The CGA>GGA (p.G1093R)
switch in the PTCH1 gene introduces the substitution of a highly
conserved glycine residue located in the 10th transmembrane region
of the PTCH protein. This region was previously described in
familiar KCOT patients (14)
showing heterozygosity at nucleotide c.3277, which differs from the
proband in being a G>A transversion and not a G>C transition,
although in both cases, the result was a G to R substitution at
amino acid position 1093. The web-based software applications
PolyPhen, SIFT and PMUT were used to predict the potential
functional and structural effects of the G1093R missense variant.
All predictions revealed that this variant is pathogenetic.
Discussion
The identification of a germline missense mutation
c.3277G>C (p.G1093R) in the PTCH1 sequence of a NBCCS patient
suggests a new hypothesis in the genetic disease process. A
complete clinical examination supported by histological and
radiological findings in the presence of related patients confirmed
the diagnosis of NBCCS. Therefore, this family may be considered
affected by NBCCS with autosomal dominant inheritance. The missense
germline mutation c.3277G>C (p.G1093R) in the PTCH1 gene,
located at chromosome 9q22.3, has only been associated with
non-syndromic KCOTs (14) and
sporadic NBCCS (16).
This particular mutation has previously been
reported in five patients from a Chinese family presenting with
only signs of KCOTs and no other major or minor criteria of NBCCS.
In this study, the mutation c.3277G>C (p.G1093R) was
considered to be causative and indicative of non-syndromic KCOTs as
it was identical in all patients in the same family following DNA
sequence analysis of all 23 exons of the PTCH1 gene (14). The same germline mutation of the
PTCH1 gene has previously only been reported in a French patient
with sporadic NBCCS (16).
Normally, the PTCH1 gene codes for a protein that is
a 12-pass-transmembrane receptor for Sonic Hedgehog and other
Hedgehog proteins for repressing the functions of the signaling
effector Smoothened, a member of the seven-transmembrane receptor
superfamily. The Hedgehog (HH) signaling pathway is pivotal in
patterning, morphogenesis and growth in numerous tissues, including
teeth (17–19). It is also known that HH signaling
regulates the proliferation of distinct cell types via the direct
activation of genes that are involved in cell cycle progression
(20). Therefore, it is plausible
that the increased proliferative activity observed in KCOTs,
compared with other odontogenic cysts, is associated with an
uncontrolled HH activity pathway (7).
Notably, this study reports an isolated missense
mutation c.3277G>C (p.G1093R) of a single amino acid
located in the 10th transmembrane region of the PTCH protein that
causes one highly conserved glycine residue transit to arginine on
the 10th transmembrane region of the PTCH protein. This glycine
residue is highly conserved among humans, mice, rats, chicks and
zebrafish, suggesting that it is functionally significant (14).
In line with previous studies, no correlation
between the position of the germline mutation and the observed
phenotype in NBCCS patients was detected (8,21) and
no genotype-phenotype correlation was found in the patients. The
lack of a genotype-phenotype correlation is known for other genes
involved in familial cancer (neurofibromatosis type 1, Muir-Torre
syndrome) for which identical mutations did not cause similar
clinical features in related and unrelated patients (22,23).
This suggests that additional factors are involved in the
development of clinical symptoms in NBCCS.
It would be useful to observe whether this amino
acid is necessary for the interaction of Hedgehog with its PTCH
receptor. However, no mutagenesis data or binding studies are
available to document the effects of this mutation.
At present, no founder effect of PTCH1 gene mutation
has been described in the literature and the present study results
did not have any reason to support a founder effect. Certain
mutations in different exons have been reported twice in the
literature without knowledge of recurrence (5,8,21). As
yet, no conclusive evidence for a genotype-phenotype correlation in
Gorlin syndrome has been demonstrated and there is adequate
variation in single families to suggest that environmental exposure
and modifier genes justify much of the variation.
The considerable inter- and intra-familial
variability in a number of genetic diseases is likely to result
from the action of modifier genes. Increasing evidence now suggests
that the manifestations of a number of genetic disorders are
influenced by modifying genes distinct from the disease locus.
Specific modifying loci have also been identified. For example, two
or more modifiers of the phenotype of the Multiple intestinal
neoplasia (Min) mouse model of familial adenomatous polyposis (FAP)
are known to exist (24). The type
and position of a mutation have been shown to affect phenotype for
a number of tumor-prone disorders, including neurofibromatosis type
2 (NF2) (25,26) and FAP (27).
This phenotypic diversity strongly suggests that the
final phenotypic outcome is modified by epigenetic factors,
including ethnicity and environmental factors. Among these,
ultraviolet (UV) light is one of the most high-risk factors for BCC
development in NBCCS (28). The
increased frequency of BCCs in Caucasian (80%) compared with
African-American individuals (38%) has been reported in other
studies (29,30) and the lower frequency of BCCs in the
African-American group may be associated with increased protection
from UV light due to skin pigmentation (31).
To the best of our knowledge, this is the first
study reporting a correlation between the missense mutation
c.3277G>C (p.G1093R) and NBCCS-associated phenotype with
the presence of multiple KCOTs, suggesting that a missense mutation
induces different clinical phenotypes. The identification of a new
mutation in NBCCS may facilitate diagnostic testing for families
and for carrier parents, siblings and other family members. An
accurate and close surveillance for dermatological and
oropharyngeal lesions as well as for the risk of malignancy
development is mandatory in these individuals.
References
|
1
|
Gorlin RJ: Nevoid basal cell carcinoma
syndrome. Dermatol Clin. 13:113–125. 1995.PubMed/NCBI
|
|
2
|
Evans DG, Ladusans EJ, Rimmer S, Burnell
LD, Thakker N and Farndon PA: Complications of the naevoid basal
cell carcinoma syndrome: results of a population based study. J Med
Genet. 30:460–464. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pastorino L, Cusano R, Nasti S, et al:
Molecular characterization of Italian nevoid basal cell carcinoma
syndrome patients. Hum Mutat. 25:322–323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stone DM, Hynes M, Armanini M, et al: The
tumour-suppressor gene patched encodes a candidate receptor for
Sonic hedgehog. Nature. 384:129–134. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hahn H, Christiansen J, Wicking C,
Zaphiropoulos PG, Chidambaram A, Gerrard B, et al: A mammalian
patched homolog is expressed in target tissues of sonic hedgehog
and maps to a region associated with developmental abnormalities. J
Biol Chem. 271:12125–12128. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johnson RL, Rothman AL, Xie J, et al:
Human homolog of patched, a candidate gene for the basal cell nevus
syndrome. Science. 272:1668–1671. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diniz MG, Borges ER, Guimarães ALS, et al:
PTCH1 isoforms in odontogenic keratocysts. Oral Oncol. 45:291–295.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wicking C, Shanley S, Smyth I, et al: Most
germ-line mutations in the nevoid basal cell carcinoma syndrome
lead to a premature termination of the PATCHED protein, and no
genotype-phenotype correlations are evident. Am J Hum Genet.
60:21–26. 1997.
|
|
9
|
Barreto DC, Gomez RS, Bale AE, Boson WL
and De Marco L: PTCH gene mutations in odontogenic keratocysts. J
Dent Res. 79:1418–1422. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Woolgar JA, Rippin JW and Browne RM: The
odontogenic keratocyst and its occurrence in the nevoid basal cell
carcinoma syndrome. Oral Surg Oral Med Oral Pathol. 64:727–730.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lench NJ, High AS, Markham AF, Hume WJ and
Robinson PA: Investigation of chromosome 9q22.3-q31 DNA marker loss
in odontogenic keratocysts. Eur J Cancer B Oral Oncol. 32:202–206.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Levanat S, Gorlin RJ, Fallet S, Johnson
DR, Fantasia JE and Bale AE: A two-hit model for developmental
defects in Gorlin syndrome. Nat Genet. 12:85–87. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lench NJ, Telford EA, High AS, Markham AF,
Wicking C and Wainwright BJ: Characterization of human patched germ
line mutations in naevoid basal cell carcinoma syndrome. Hum Genet.
100:497–502. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Lu Y, Shen G and Chen W: One
germline mutation of PTCH gene in a Chinese family with
non-syndromic keratocystic odontogenic tumours. Int J Oral
Maxillofac Surg. 40:829–833. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kimonis VE, Goldstein AM, Pastakia B, et
al: Clinical manifestations in 105 persons with nevoid basal cell
carcinoma syndrome. Am J Med Genet. 69:299–308. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pruvost-Balland C, Gorry P, Boutet N, et
al: Clinical and genetic study in 22 patients with basal cell nevus
syndrome. Ann Dermatol Venereol. 133:117–123. 2006.PubMed/NCBI
|
|
17
|
Dassule HR, Lewis P, Bei M, Maas R and
McMahon AP: Sonic hedgehog regulates growth and morphogenesis of
the tooth. Development. 127:4775–4785. 2000.PubMed/NCBI
|
|
18
|
Oro AE and Higgins K: Hair cycle
regulation of Hedgehog signal reception. Dev Biol. 255:238–248.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hebrok M, Kim SK, St Jacques B, McMahon AP
and Melton DA: Regulation of pancreas development by hedgehog
signaling. Development. 127:4905–4913. 2000.PubMed/NCBI
|
|
20
|
Duman-Scheel M, Weng L, Xin S and Du W:
Hedgehog regulates cell growth and proliferation by inducing Cyclin
D and Cyclin E. Nature. 417:299–304. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boutet N, Bignon YJ, Drouin-Garraud V, et
al: Spectrum of PTCH1 mutations in French patients with Gorlin
syndrome. J Invest Dermatol. 121:478–481. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ponti G, Losi L, Martorana D, et al:
Clinicopathological and biomolecular findings in Italian patients
with multiple cutaneous neurofibromas. Hered Cancer Clin Pract.
9:62011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ponti G, Losi L, Pedroni M, et al: Value
of MLH1 and MSH2 mutations in the appearance of Muir-Torre syndrome
phenotype in HNPCC patients presenting sebaceous gland tumors or
keratoacanthomas. J Invest Dermatol. 126:2302–2307. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Houlston RS and Tomlinson IP: Modifier
genes in humans: strategies for identification. Eur J Hum Genet.
6:80–88. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Evans DGR, Trueman L, Wallace A, Collins S
and Strachan T: Genotype/phenotype correlations in type 2
neurofibromatosis (NF2): evidence for more severe disease
associated with truncating mutations. J Med Genet. 35:450–455.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baser ME, Friedman JM, Aeschliman D, et
al: Predictors of the risk of mortality in neurofibromatosis 2. Am
J Hum Genet. 71:715–723. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Davies DR, Armstrong JG, Thakker N, et al:
Severe Gardner syndrome in families with mutations restricted to a
specific region of the APC gene. Am J Hum Genet. 57:1151–1158.
1995.PubMed/NCBI
|
|
28
|
Tanioka M, Takahashi K, Kawabata T, et al:
Germline mutations of the PTCH gene in Japanese patients with
nevoid basal cell carcinoma syndrome. Arch Dermatol Res 2005.
296:303–308. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Anderson DE, Taylor WB, Falls HF and
Davidson RT: The nevoid basal cell carcinoma syndrome. Am J Hum
Genet. 19:12–22. 1967.PubMed/NCBI
|
|
30
|
Goldstein AM, Bale SJ, Peck GL and Di
Giovanna JJ: Sun exposure and basal cell carcinomas in the nevoid
basal cell carcinoma syndrome. J Am Acad Dermatol. 29:34–41. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Goldstein AM, Pastakia B, Di Giovanna JJ,
et al: Clinical findings in two African-American families with the
nevoid basal cell carcinoma syndrome (NBCC). Am J Med Genet.
50:272–281. 1994. View Article : Google Scholar : PubMed/NCBI
|