Introduction
Cytotoxic nucleoside analogues are widely used in
cancer chemotherapy for colorectal, breast, head and neck,
non-small cell lung, pancreatic cancer and leukaemia. Most
antitumour 2′-deoxycytidine analogues, such as gemcitabine (GEM)
and cytosine arabinoside (Ara-C), have common antitumour mechanisms
and metabolic pathways (1). These
nucleosides must first be transported into the cell via a
nucleoside transporter. The most active uptake has been found to be
via the human equilibrative nucleoside transporter 1 (hENT1) and
efflux has been found to occur via the ATP-binding cassette (ABC)
transporter (2–4). Once inside a cell, these nucleosides
are phosphorylated by deoxycytidine kinase (dCK) to the active
triphosphate form (5), which is
then most likely incorporated into DNA. The active diphosphate
metabolite inhibits DNA synthesis indirectly through the inhibition
of ribonucleoside reductase (RR). RR has two subunits (RRM1 and
RRM2) and converts ribonucleoside 5′-diphosphates to the
deoxyribonucleotide 5′-diphosphates that are essential for DNA
synthesis. Therefore, the mechanism of sensitivity and/or
resistance to 2′-deoxycytidine analogues is likely to be
multifactorial, involving decreased intracellular accumulation and
the alteration of metabolism by the aforementioned molecules
(1–7).
Pemetrexed (MTA) is a multitarget antifolate
cytotoxic agent available for use in the treatment of non-small
cell lung cancer (NSCLC) and malignant pleural mesothelioma
(8,9). We previously reported that MTA
pretreatment altered the expression of dCK, resulting in the
enhanced cytotoxicity of GEM in GEM-resistant NSCLC cells (4). In the present study, we have further
examined the molecules involved in the synergistic interaction of
MTA and 2′-deoxycytidine analogues.
Materials and methods
Cell lines and chemicals
The human erythroleukaemia cell line K562 and the
myeloid leukaemia cell line HL60 were used in this study. Cells
from the MTA-resistant human small cell lung carcinoma cell lines
PC6/MTA-0.4 and PC6/MTA-1.6 were established from parental PC-6
cells as described previously (10,11).
PC6/MTA-0.4 cells were cultured with 0.4 μM MTA and
PC6/MTA-1.6 cells with 1.6 μM MTA. The 5-fluorouracil
(5-FU)-resistant subline PC-6/FU23-26, selected from PC-6 human
small cell lung cancer cells as described previously, was used as a
control (12). The cells were
cultured in RPMI-1640 (or, for A549, in Dulbecco’s modified Eagle’s
medium) supplemented with 10% heat-inactivated FBS and 1% (v/w)
penicillin/streptomycin in a humidified chamber (37°C, 5%
CO2). GEM and MTA were provided by Eli Lilly
Pharmaceuticals (Indianapolis, IN, USA). Ara-C was purchased from
Wako Pure Chemical Industries (Osaka, Japan).
Total RNA extraction and quantitative
RT-PCR
Total RNA was extracted using an RNeasy Mini kit
(Qiagen, Hilden, Germany). Quantitative real-time RT-PCR was
performed in a volume of 20 μl with a Taqman One-Step RT-PCR
Master Mix Reagents kit (Applied Biosystems, Foster City, CA, USA)
using the StepOnePlus Real-Time PCR System (Applied Biosystems)
according to the manufacturer’s instructions. Melting curve
analysis was used to control for the specificity of the
amplification products. The number of transcripts was calculated
from a standard curve obtained by plotting the input of four
different known transcript concentrations versus the PCR cycle
number at which the detected fluorescence intensity reached a fixed
value. The PCR program consisted of 45 cycles of 94°C for 15 sec
and 60°C for 1 min. The experiment was performed in triplicate. The
primer and Taqman probe sets (Taqman Gene Expression Assays,
Inventoried) for dCK (Hs00176127_m1), hENT1 (Hs01085706_m1),
multidrug resistance protein 5 (ABCC5; Hs00981087_m1), RRM1
(Hs00168784_m1) and glyceraldehyde-3-phosphate dehydrogenase
(GAPDH; Hs99999905_m1) were purchased from Applied Biosystems
(sequences not disclosed). Expression was normalised to the
housekeeping gene GAPDH.
Concentration of GEM and Ara-C for cell
survival
Cells were cultured at 5,000 cells/well in 96-well
tissue culture plates. To assess cell viability, ten-fold dilutions
of the anticancer drug were added stepwise 2 h after plating, and
the cultures were incubated at 37°C for 96 h. At the end of the
culture period, 20 μl of MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-
(4-sulfophenyl)-2H-tetrazolium, inner salt] solution (CellTiter 96®
AQueous One Solution Cell Proliferation Assay, Promega, Madison,
WI, USA) was added, the cells were incubated for a further 4 h and
the absorbance was measured at 490 nm using an ELISA plate reader.
Mean values were calculated from three independent experiments
performed in quadruplicate. Chemosensitivity is expressed as the
drug concentration for IC50, determined from the
concentration-effect correlation using Graph Pad Prism version 4
(GraphPad Software, San Diego, CA, USA).
Statistical analysis
The differences in cell viability between samples
were evaluated using the Student’s unpaired t-test. The level of
significance was set at 5%, using a two-sided analysis.
Results
Gene expression levels of dCK, hENT1,
ABCC5 and RRM1 in leukaemia cells
Previously, we examined dCK, hENT1, ABCC5 and RRM1
expression in GEM-resistant NSCLC cells, and found that alteration
of these molecules was associated with sensitivity and/or
resistance to GEM (3,4,7).
Therefore, to examine the differential expression of dCK, hENT1,
ABCC5 and RRM1 for Ara-C sensitivity, we used two leukaemia cell
lines: Ara-C-resistant erythroleukaemia K562 cells and
Ara-C-sensitive myeloid leukaemia HL60 cells. We found that the
levels of expression of ABCC5 and RRM1 were lower in HL60 cells
than in K562 cells. By contrast, the expression levels of hENT1 and
dCK were higher in HL60 cells than in K562 cells (Fig. 1).
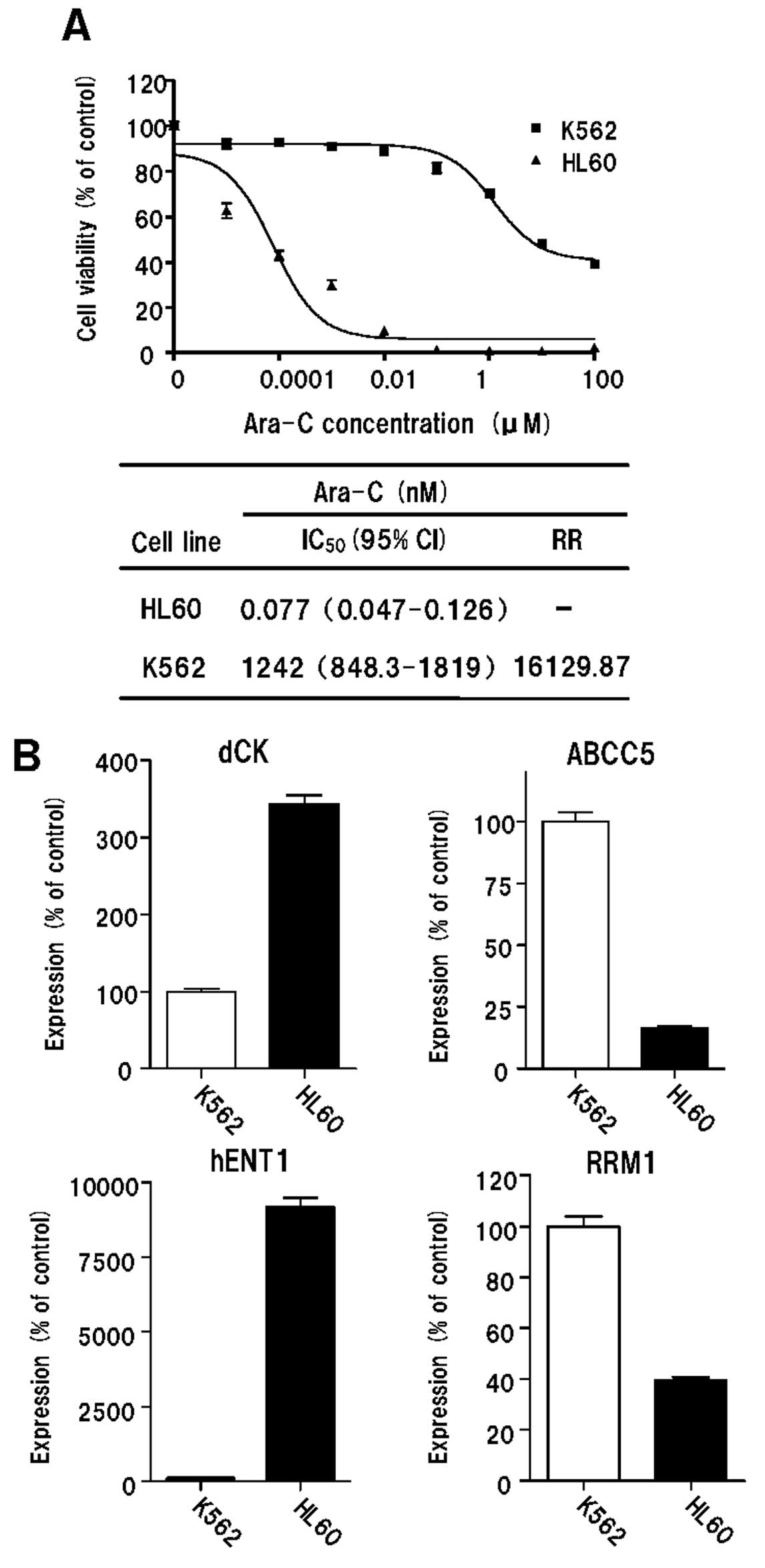 | Figure 1Cytotoxicity of Ara-C and expression
levels of dCK, ABCC5, hENT1 and RRM1 genes in K562 and HL60 cells.
(A) Cytotoxicity of Ara-C in K562 and HL60 cells. (B) Expression
levels of dCK, ABCC5, hENT1 and RRM1 in K562 and HL60 cells genes,
as determined by real-time PCR. Ara-C, cytosine arabinoside; dCK,
deoxycytidine kinase; ABCC5, multidrug resistance protein 5; hENT1,
human equilibrative nucleoside transporter 1; RRM1, ribonucleoside
reductase subunit M1; IC50, concentration for 50% cell
survival; CI, confidence interval; RR, resistance rate. |
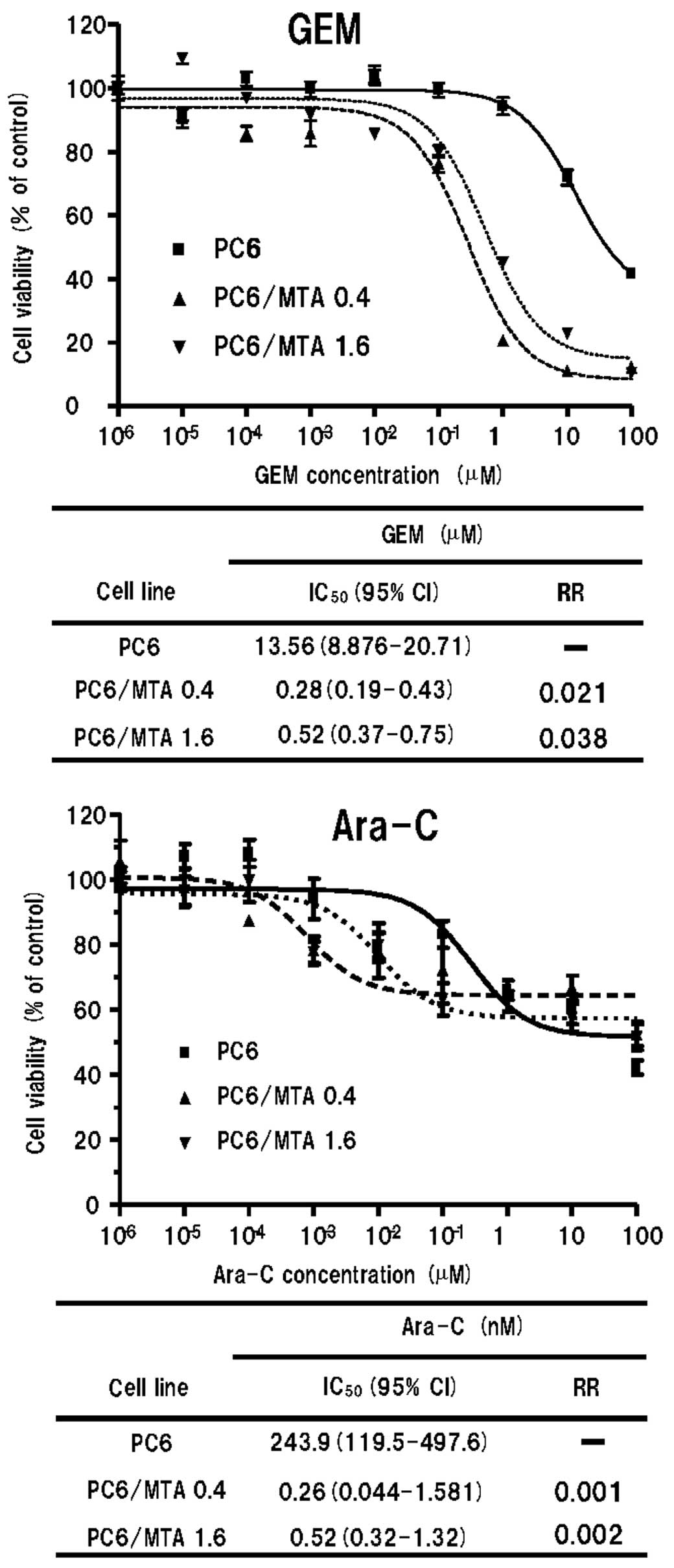
Sensitivity to GEM and Ara-C in
MTA-resistant cells
The sensitivities of parental and MTA-resistant
cells to GEM and Ara-C are summarised in Fig. 2. Both MTA-resistant cell lines,
PC6/MTA-0.4 and PC6/MTA-1.6, were more sensitive to GEM and Ara-C
than the parental PC6 cells.
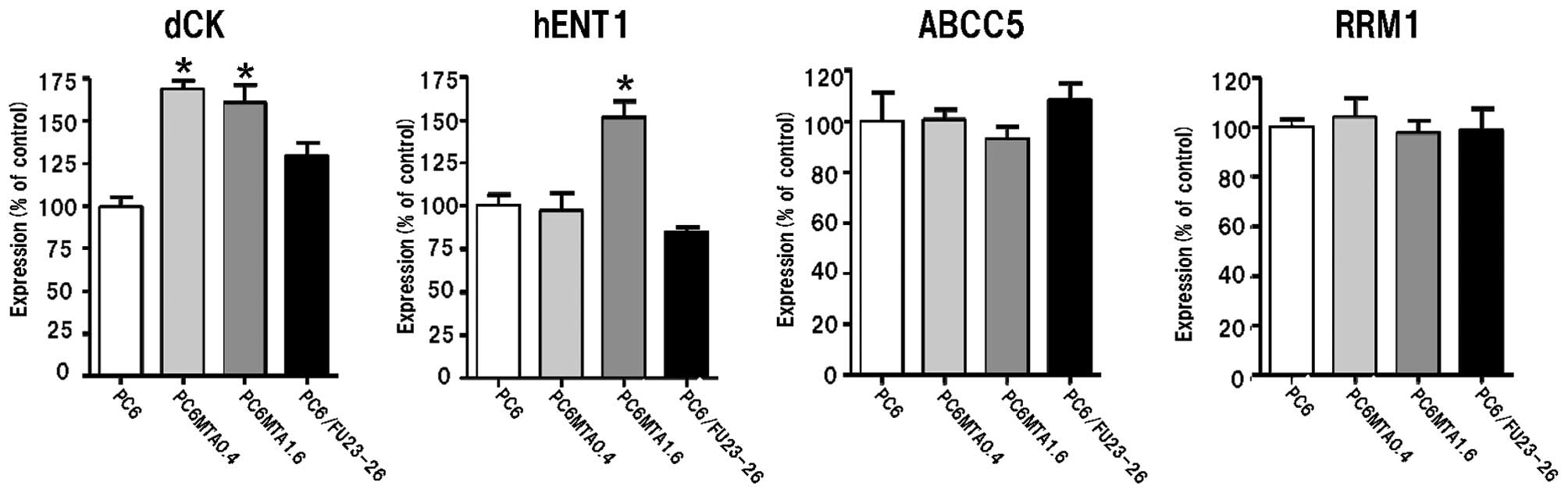
Gene expression levels of dCK, hENT1,
ABCC5 and RRM1 in MTA-resistant cells
We examined the level of gene expression of dCK,
hENT1, ABCC5 and RRM1 in parental and MTA-resistant cells. As shown
in Fig. 3, the expression levels of
hENT1 and dCK in MTA-resistant cells were significantly higher than
in the parental cells (P<0.05). We used 5-FU-resistant
PC6/FU23-26 cells as a control as the pyrimidine analogue 5-FU,
while also an antimetabolite drug, differs from 2′-deoxycytidine
analogues in that it requires intracellular conversion (12). The expression levels of hENT1 and
dCK in PC6/FU23-26 cells were not changed compared with PC6 cells.
These results indicate that the alteration of these molecules
affects sensitivity to GEM and Ara-C in MTA-resistant cells.
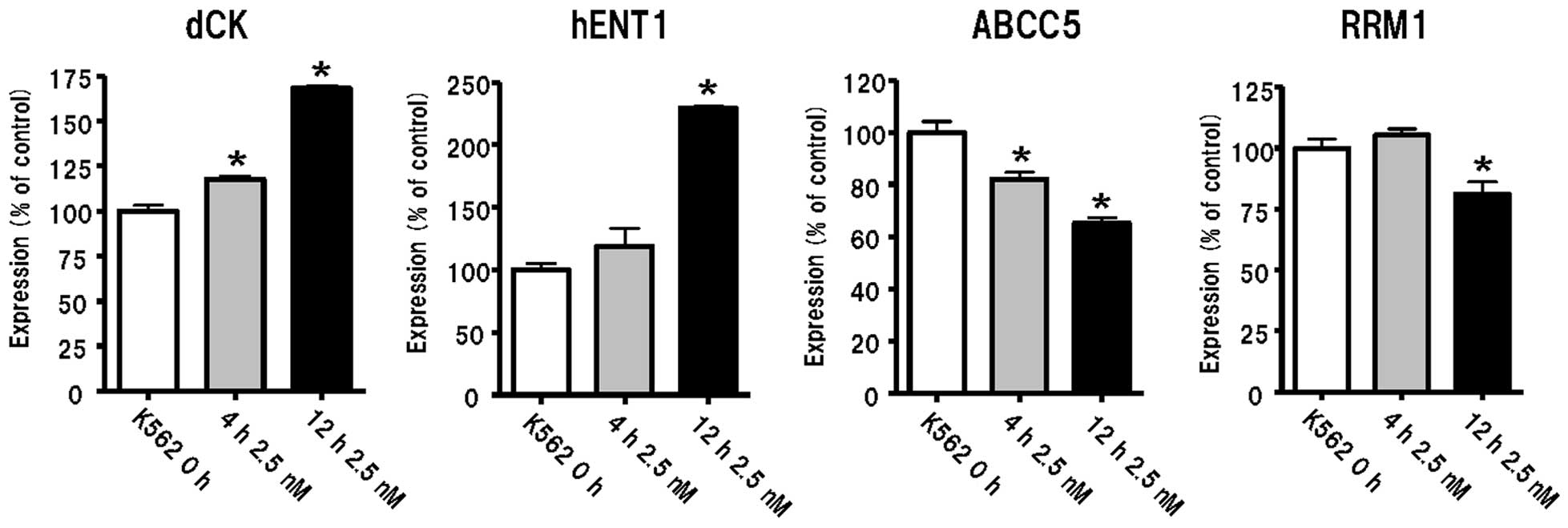
Modification of Ara-C cytotoxicity by MTA
treatment
We examined gene expression levels of dCK, hENT1,
ABCC5 and RRM1 in K562 cells following treatment with 2.5 nM MTA
for 4 and 12 h. We found increased levels of expression of hENT1
and dCK and decreased expression of ABCC5 and RRM1 (P<0.05,
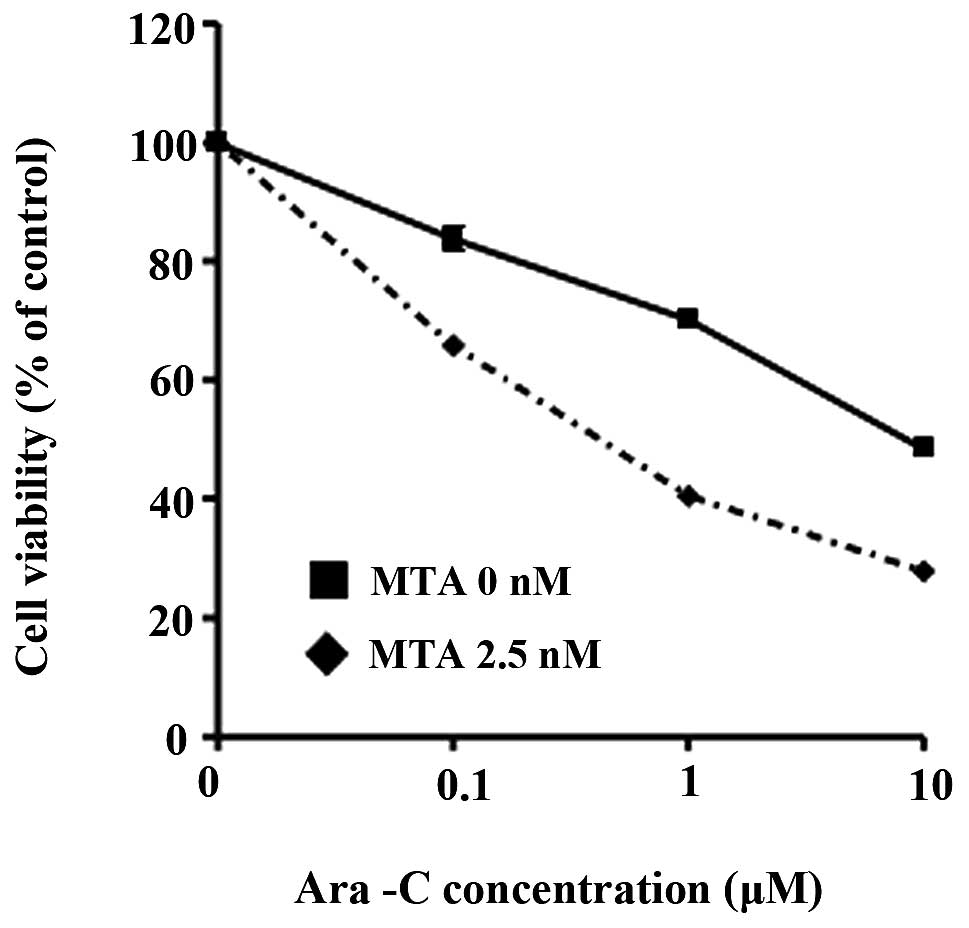
Fig. 4). We also found that
pretreatment with 2.5 nM of MTA for 12 h altered the cytotoxicity
of Ara-C in K562 cells (Fig.
5).
Discussion
We found that the gene expression levels of dCK,
hENT1, ABCC5 and RRM1 are different in Ara-C-sensitive and
-resistant leukaemia cells.
We previously reported that ABCC5, hENT1, dCK and
RRM1 expression levels are associated with sensitivity and/or
resistance to GEM (4). hENT1 and
ABCC5 are associated with drug uptake and efflux, meaning that
increased expression of hENT1 and decreased expression of ABCC5 may
result in increased intracellular concentration of cytotoxic
nucleoside analogues, which is an important factor influencing
their cytotoxicity. By contrast, dCK mediates the rate-limiting
step, which is the first phosphorylation, in the process that
converts anticancer nucleoside analogues to their active
triphosphate form (2). Increased
RRM1 increases the size of deoxynucleoside triphosphate (dNTP)
pools that competitively inhibit the incorporation of the
triphosphate cytotoxic nucleoside analogue into DNA (2). These increased dNTP pools further
downregulate the activity of dCK via a negative-feedback pathway.
The diphosphate forms of cytotoxic nucleoside analogues inhibit
RRM1, resulting in a decrease in dNTP pools (6). Therefore, increased expression of dCK
and decreased expression of RRM1 may result in the increased
activation of anticancer nucleoside analogues. Alteration of the
expression of dCK, hENT1, ABCC5 and RRM1 affects the intracellular
concentration and activation of GEM and Ara-C.
Previous studies have revealed that the
downregulation of dCK increased the resistance of human leukaemia
cells to cladribine and clofarabine (13,14).
We previously showed that treatment with MTA enhances dCK
expression, concomitant with altering the cytotoxicity of GEM
(4). Indeed, the clinical
administration of MTA followed by GEM met the protocol-defined
efficacy criteria, and was less toxic than administering GEM
followed by MTA (15). Therefore,
we investigated the effect of treatment with MTA on Ara-C
sensitivity. We found that MTA-resistant cells were more sensitive
to GEM and Ara-C concomitant with increased expression of dCK and
hENT1. Further, pretreatment with MTA enhanced not only dCK
expression, but also ABCC5 and hENT1 expression concomitant with
altering the sensitivity to Ara-C in Ara-C-resistant K562 cells.
Thus, the combination of Ara-C and MTA exhibits order-dependent
synergistic cytotoxic activity, suggesting the efficacy of
gene-expression modulation by chemotherapy combinations. Ara-C is
currently used to treat haematological malignancies, especially as
induction chemotherapy for acute myeloid leukaemia (AML) (16). High-dose Ara-C is also used to treat
relapsed or refractory AML, and induction chemotherapy with
high-dose Ara-C has been shown to improve overall survival in
clinical trials (17). Although
high-dose Ara-C is most effective for relapsed or refractory AML,
toxicity, especially non-haematological toxicity such as severe
infection, emesis and central nervous system toxicity, is common
(16,17). While MTA has not been approved for
haematological malignancies, its toxicity is mild when supplemented
with folic acid and vitamin B12 (18). Our in vitro study shows the
efficacy of the combination of MTA and Ara-C, suggesting a new
treatment option for AML chemotherapy.
We found that the mechanisms of the synergistic
interaction of MTA and cytotoxic nucleoside analogues are
multifactorial. Future studies should examine whether these factors
may be used as predictive markers for cytotoxic nucleoside
analogues.
Acknowledgements
This study was supported by a Grant-in-Aid for
Scientific Research (c) from the Japan Society for the Promotion of
Science (MEXT/JSPSKAKENHI23591156).
References
|
1
|
Galmarini CM, Mackey JR and Dumontet C:
Nucleoside analogues and nucleobases in cancer treatment. Lancet
Oncol. 3:415–424. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heinemann V, Schulz L, Issels RD and
Plunkett W: Gemcitabine: a modulator of intracellular nucleotide
and deoxynucleotide metabolism. Semin Oncol. 22(4 Suppl 11): 11–18.
1995.PubMed/NCBI
|
|
3
|
Achiwa H, Oguri T, Sato S, Maeda H, Niimi
T and Ueda R: Determinants of sensitivity and resistance to
gemcitabine: the roles of human equilibrative nucleoside
transporter 1 and deoxycytidine kinase in non-small cell lung
cancer. Cancer Sci. 95:753–757. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oguri T, Achiwa H, Sato S, et al: The
determinants of sensitivity and acquired resistance to gemcitabine
differ in non-small-cell lung cancer: a role of ABCC5 in
gemcitabine sensitivity. Mol Cancer Ther. 5:1800–1806. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bergman AM, Pinedo HM and Peters GJ:
Determinants of resistance to 2′,2′-difluorodeoxycytidine
(gemcitabine). Drug Resist Updat. 5:19–33. 2002.
|
|
6
|
Davidson JD, Ma L, Flagella M, Geeganage
S, Gelbert LM and Slapak CA: An increase in the expression of
ribonucleotide reductase large subunit 1 is associated with
gemcitabine resistance in non-small cell lung cancer cell lines.
Cancer Res. 64:3761–3766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oguri T, Achiwa H, Muramatsu H, et al: The
absence of human equilibrative nucleoside transporter 1 expression
predicts nonresponse to gemcitabine-containing chemotherapy in
non-small cell lung cancer. Cancer Lett. 256:112–119. 2007.
View Article : Google Scholar
|
|
8
|
Hazarika M, White RM, Johnson JR and
Pazdur R: FDA drug approval summaries: pemetrexed (Alimta).
Oncologist. 9:482–488. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cohen MH, Justice R and Pazdur R: Approval
summary: pemetrexed in the initial treatment of advanced/metastatic
non-small cell lung cancer. Oncologist. 14:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ozasa H, Oguri T, Uemura T, et al:
Significance of thymidylate synthase for resistance to pemetrexed
in lung cancer. Cancer Sci. 101:161–166. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Uemura T, Oguri T, Ozasa H, et al:
ABCC11/MRP8 confers pemetrexed resistance in lung cancer. Cancer
Sci. 101:2404–2410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oguri T, Bessho Y, Achiwa H, et al:
MRP8/ABCC11 directly confers resistance to 5-fluorouracil. Mol
Cancer Ther. 6:122–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Månsson E, Flordal E, Liliemark J, et al:
Down-regulation of deoxycytidine kinase in human leukemic cell
lines resistant to cladribine and clofarabine and increased
ribonucleotide reductase activity contributes to fludarabine
resistance. Biochem Pharmacol. 65:237–247. 2003.
|
|
14
|
Månsson E, Spasokoukotskaja T, Sällström
J, Eriksson S and Albertioni F: Molecular and biochemical
mechanisms of fludarabine and cladribine resistance in a human
promyelocytic cell line. Cancer Res. 59:5956–5963. 1999.PubMed/NCBI
|
|
15
|
Ma CX, Nair S, Thomas S, Mandrekar SJ, et
al; North Central Cancer Treatment Group; Mayo Clinic; Eli Lilly
& Company. Randomized phase II trial of three schedules of
pemetrexed and gemcitabine as front-line therapy for advanced
non-small-cell lung cancer. J Clin Oncol. 23:5929–5937. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tallman MS, Gilliland DG and Rowe JM: Drug
therapy for acute myeloid leukemia. Blood. 106:1154–1163. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kern W and Estey EH: High-dose cytosine
arabinoside in the treatment of acute myeloid leukemia: Review of
three randomized trials. Cancer. 107:116–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohe Y, Ichinose Y, Nakagawa K, et al:
Efficacy and safety of two doses of pemetrexed supplemented with
folic acid and vitamin B12 in previously treated patients with
non-small cell lung cancer. Clin Cancer Res. 14:4206–4212. 2008.
View Article : Google Scholar : PubMed/NCBI
|