Contents
Introduction
CCRK: A novel CAK in mammalian cells
The role of CCRK as CAK in carcinogenesis
The role of CCRK as a novel oncoprotein in
carcinogenesis
CCRK as a promising target for cancer therapy
Conclusion
Introduction
Dysregulation of the cell cycle components may lead
to tumor formation. The formation of tumors occurs when genes such
as CDK, CDK-activating kinase (CAK), RB and p53 mutate, causing the
cell to multiply uncontrollably (1–3).
Regulation of the cell cycle progression appears to be achieved
principally by cyclins and CDK activity at the G1/S and G2/M phase
transitions (4). The mid-G1 phase
is characterized by the interaction between cyclin D1 and cdk4/6
(5). Another important complex at
the G1/S boundary is that of Cdk2 and cyclin E (cyc E) (6). This complex hyperphosphorylates the
retinoblastoma protein (pRb) and its family members (7). For total activation, a CDK must be
binding to the subunits of cyclin, and then phosphorylated by a CAK
within the activation segment or T-loop (8). Cells lacking CAKs are incapable of
growth, whereas cells with a reduced level of CAKs exhibited
retarded growth compared to control cells (9,10).
Additionally, a specific inhibition of CAKs was found to promote
tumor cell death (11,12).
Cell proliferation is achieved through the
transition of cells from G0/G1 arrest into the active cell cycle.
Although progression through the cell cycle is controlled by the
regulatory proteins mentioned above, overall proliferation is
modulated by signaling pathways. The growth signal transduction is
disrupted in almost all tumor types, as in the case of the Wnt
pathway, which regulates the cell cycle mechanism to control cell
proliferation and tumorigenesis (13,14).
Aberrant activation of the Wnt pathway occurs in several human
malignancies such as hepatocellular carcinoma (HCC),
gastrointestinal cancer and breast cancer (15,16).
Recent studies have shown that the cell cycle-related kinase (CCRK)
exhibits strong oncogenic properties in HCC by activating
Wnt/β-catenin signaling and in turn upregulating the expression of
β-catenin downstream targets (17).
CCRK, also known as CDK20 or p42, first reported in
HeLa cells by Kaldis and Solomon in 2000 (18), is a newly identified 42 kDa protein
containing all 11 conserved subdomains characteristic of
serine/threonine protein kinase (19). CCRK has sequence homology to both
Cak1p and CDK7 groups of CAKs (20). Currently, CCRK is known for its role
in CAK function, independently of Cdk7 complexes. CCRK is capable
of phosphorylating CDK2 on Thr-160 in vitro, an intrinsic
kinase activity of CDK2 that is not required for this
phosphorylation (8,11,20–22).
Recent studies have indicated that CCRK is widely expressed in cell
lines originating from a variety of tumor tissues, and CCRK
suppression would cause cell cycle arrest at the G0–G1 phase by
decreasing the phosphorylation of Cdk2/Rb (11,17,21,23).
In addition, CCRK is overexpressed in a number of human
malignancies. Mouse models of CCRK functions have provided data
suggesting that the overexpression of CCRK promotes tumorigenicity
(17,23). Moreover, CCRK knockdown decreased
the expression of cyclins and β-catenin in various types of human
cancer. Therefore, CCRK phosphorylates Cdk2 and interacts with
other proteins to act as an oncoprotein (17,23).
CCRK is involved in a wide array of cell signaling pathways
associated with cell proliferation, which is closely linked to the
genesis and evolution of cancer (17). These findings suggest that CCRK is a
promising target of cancer therapy as a novel CAK or oncoprotein.
In this review, we focused on the compilation of information
available thus far regarding the significance of CCRK in
carcinogenesis for future investigations.
CCRK: A novel CAK in mammalian cells
Cdk activation requires CAK phosphorylation of the
cylin-Cdk complex on the threonine residue in the activation
segment. Several lines of evidence support the role of CAK as an
important cell cycle regulator, becoming a target for the design of
inhibitors to regulate the cell cycle, especially in diseases such
as cancer (24–27). In addition to activating Cdks, CAK
regulates transcription (28). CAK
associated with TFIIH phosphorylates proteins involved in
transcription, including RNA polymerase II (29,30).
Moreover, CAK plays a role in DNA damage response. The activity of
CAK associated with TFIIH decreases when DNA is damaged by UV
irradiation (29). Inhibition of
CAK prevents cell cycle progression, a mechanism ensuring the
fidelity of chromosome transmission. The Cdk7 complex is the first
and best-characterised CAK that is activated throughout the cell
cycle (31). The Cdks1, 2, 4, 5 and
6 alone are inactive and require both association with a cyclin and
phosphorylation on a conserved threonine residue by Cdk7 complex
(32–36). However, the existence of an
additional CAK, other than CDK7, has also been suggested. Kaldis
and Solomon (18) biochemically
enriched a ‘small CAK’ activity in cervical carcinoma HeLa cells.
This small CAK activity is distinct in size from CDK7 activity, the
former peaks at 42 kDa whereas the latter peaks at 140 kDa
(18). Transforming growth factor
(TGF) treatment in osteosarcoma U2OS cells resulted in low CDK2
Thr-160 phosphorylation without affecting CDK7 activity (37). Moreover, in D. melanogaster,
inactivation of the CDK7 homolog with null and
temperature-sensitive mutations showed that CDK7 is required for
the activation of CDC2-cyclin A and CDC2-cyclin B complexes, but
not for CDK2-cyc E complexes (12).
CCRK, the ‘small CAK’ delineated by Kaldis and
Solomon (18), is a novel CAK based
on: i) limited homology to Cdk7 and related kinases, with CCRK
being a 346-amino acid protein with a molecular weight of 42 kDa
that shares 43% sequence identity with CDK7. The ATP analog FSBA
did not inhibit CCRK activity whereas it completely inactivated
Cdk7 (19,20); ii) a modest level of CAK activity
in vitro albeit weaker than CDK7 (20,38).
CCRK has a substrate specificity that is different from CDK7, with
CCRK favoring CDK2 and MAK-related kinase/intestinal cell kinase
(MRK/ICK) as the substrate (20,29,38–40);
iii) a cell proliferation defect, apparently accompanied by the
decreasing phosphorylation of Cdk2 Thr-160, upon RNAi-mediated
knockdown in human cells (17,21,23). A
study by Wohlbold et al (19) demonstrated that although CCRK
supports colorectal carcinoma HCT116 cell or osteosarcoma U2OS cell
proliferation, it does not possess CAK activity against Cdk2.
Moreover, those authors found that CAK activity observed in CCRK
immuno precipitates was due to associated Cdk7 protein. The
conflicting observation may be attributed to the lack of complete
understanding of CCRK functions and difference in the cell lines.
CCRK is known to function in both a CAK and non-CAK manner in
different cell lines (11,17,22,38).
Thus, we provide the first evidence, to the best of our knowledge,
that CCRK is a novel CAK in mammalian cells.
The main subcellular localisation of CCRK is nuclear
and perinuclear regions, with a relatively low expression in the
cytoplasm (23). In human tissues,
CCRK is expressed predominantly in the brain and kidney, and to
lesser extent in the liver, heart and placenta (20). The cardiac CCRK isoform is
detectable only in heart, liver and kidney (22). In their study, Qiu et al
(22) indicated that the cardiac
CCRK expressed in the heart has been shown to promote cell
proliferation and reduce apoptosis induced by chelerythrine. CCRK
is also widely expressed in various cancer cell lines such as
glioblastoma (U87, U118, U138, U373 and SW1088), cervical
adenocarcinoma (HeLa), colorectal carcinoma (HCT116), osteogenic
sarcoma (U2OS), breast adenocarcinoma (MCF-7), ovarian carcinoma
(UACC-1598, UACC-326, OVCAR-3, HO-8910 and TOV-21G), lung
fibroblast (WI-38), myoblast (C2C12) and lymphocyte (GM08336)
(20,23). Notably, the disruption of CCRK
homeostasis is closely linked to the formation of various types of
human cancer (Table I). A high
expression level of CCRK is a strong and an independent predictor
of short overall survival in ovarian carcinoma and HCC (17,23).
Thus, CCRK expression appears to have the potential to predict
ovarian carcinoma and HCC patient clinical outcome (17). These data suggest that CCRK is
essential for the proliferation of several human malignancies.
Thus, the functions of CCRK in both a CAK and an oncoprotein manner
were delineated.
 | Table I.Overexpression of CCRK in the human
tumors investigated. |
Table I.
Overexpression of CCRK in the human
tumors investigated.
| Tumor type | No.
investigated | No. with elevated
CCRK | Refs. |
|---|
| Hepatocellular
carcinoma | 52 | 45 | (17) |
| Ovarian
carcinoma | 122 | 65 | (23) |
| Colorectal
cancer | 10 | 7 | (11) |
| Glioblastoma | 19 | 14 | (21) |
The role of CCRK as CAK in
carcinogenesis
Liu et al (20) were the first to show that CCRK has
CAK activity that regulates cell growth. In cervical adenocarcinoma
HeLa cells, the RNAi-mediated ablation of CCRK was able to inhibit
cell proliferation, cause cell cycle G1 phase arrest, decrease
pCdk2 levels and inhibit Cdk2 kinase activity (20). Similar results were observed in
human glioblastoma (21), in which
suppression of CCRK by small interfering RNA (siRNA) inhibited
glioblastoma cell growth, induced cell cycle G1 arrest and
decreased the pCdk2 level, whereas the overexpression of CCRK was
able to confer tumorigenicity to a non-tumorigenic glioblastoma
cell line, indicating that CCRK expression levels potentially
correlate with the tumorigenicity of glioblastoma cells. In another
study, An et al (11)
investigated the function of CCRK in colorectal cancer
carcinogenesis. Those authors showed that CCRK knockdown by siCCRK
or shCCRK significantly reduces cell growth and causes cell cycle
arrest at the G0–G1 phase in both LoVo and DLD1 human colorectal
cancer cell lines. In addition, cyc E-Cdk2 complexes, the important
complex at the G1/S boundary, reduced phosphorylation when CCRK was
knocked down. Moreover, siCCRK transfection markedly reduced pRb
phosphorylation. The pRb is a tumor suppressor protein that is
dysfunctional in several major cancers (41). Besides, it is worth noting that
knockdown of CCRK induced cell cycle arrest at the G2-M phase in
the LoVo cells.
The evidence mentioned above suggests that although
the overall picture of CCRK regulation remains incomplete, various
aspects can be merged to provide a perspective of the role of this
kinase cell cycle disruption. In its role as a catalyst for Cdk2
activity, CCRK provokes tumor-associated cell cycle defects to
induce unscheduled proliferation. Excess CCRK phosphorylates the
Cdk2 on Thr-160, subsequently promoting the transition from the G1
to S phase through the phosphorylation of key target proteins,
including the pRb as well as the Rb family members p130 and p107.
The hyperphosphorylated RB releases transcription factors such as
E2F that are complexed with pRb (42). In turn, E2F binds to and activates
the promoters of genes important in DNA synthesis (43). Additionally, CCRK only rarely
phosphorylates CDK2. Phosphorylation by CCRK is stimulated by the
association of CDK2 with its relevant cyclin, cyc E. After cyc E
binding, the activation segment moves towards the C-terminal lobe
exposing Thr160 for CCRK phosphorylation (43–45).
In colorectal cancer cell lines, CCRK knockdown decreases the
phosphorylation of Cdk2/Rb and reduces the expression of cyc E,
suggesting that CCRK interacts with cyc E to upregulate the
expression of the latter, enabling CCRK to phosphorylate CDK2
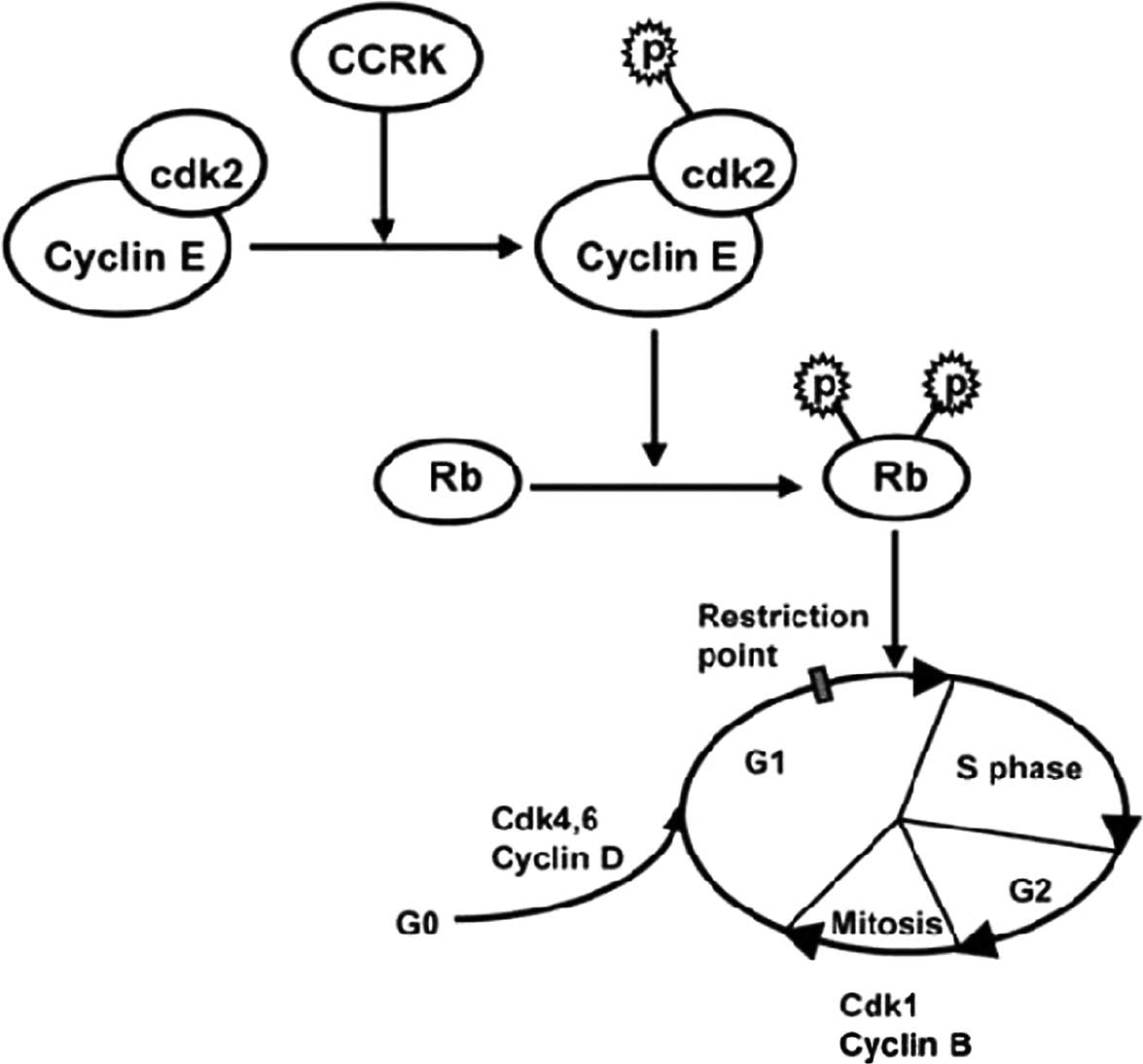
(11) (Fig. 1).
A similar regulatory mechanism has been observed
whereby CCRK activates human MAK (male germ cell-associated kinase)
and MRK/ICK by phosphorylation of the essential Thr-157 in the
T-loop (38,46). Both MAK and MRK reportedly
phosphorylate Scythe, an antiapoptotic protein required for
apoptosis and proliferation during mammalian development, in
vitro (47–49). Recent studies have shown that
MRK/MAK are new downstream targets of protein phosphatase 5 (PP5),
which plays a crucial role in biological functions and controls
almost every cell process, including gene transcription and
translation, cell-cycle progression, DNA damage response and
proliferation. Inhibition of PP5 expression leads to a marked
antiproliferative effect by activation of the p53-dependent G1
checkpoint (50,51). These findings suggest that MRK
and/or MAK play a key role in cell-cycle checkpoint control and
apoptosis in response to stress conditions, such as DNA damage.
Moreover, the phenotypic effects of silencing CCRK in malignant
cells may channel through MRK and/or MAK (52). Detailed investigations are essential
to confirm the existence of the relationship between CCRK and
MRK/MAK.
Role of CCRK as a novel oncoprotein in
carcinogenesis
In addition to phosphorylating and activating CDK2,
CCRK possesses an alternative mechanism for CCRK-induced cell cycle
progression and cell proliferation. Previous studies have shown
that CCRK is involved in a wide array of cell signaling pathways
such as the Wnt and androgen receptor (AR) signaling pathways. The
Wnt signaling pathway is a network of proteins best known for its
role in embryogenesis and cancer (13,14).
Aberrant activation of the Wnt signaling pathway occurs in several
human cancers such as HCC, gastrointestinal cancer and breast
cancer (15,16). Advances in molecular research have
demonstrated that AR signaling is involved in key cell processes
such as proliferation and apoptosis. Accumulating evidence has
shown that AR is a master regulator in the G1/S phase progression
in cancer cells and the crosstalk between this ligand-activated
transcription factor and cell cycle pathways (53,54).
It was reported that CCRK links the AR and
Wnt/β-catenin/TCF cascades and provides a mechanistic basis for the
aberrant β-catenin activation in human HCCs (17). β-catenin is a subunit of the
cadherin protein complex and has been suggested as the most
important integral component in the Wnt signaling pathway (55,56).
Upon activation, β-catenin is able to escape from the destruction
complex comprising APC, Axin and glycogen synthase kinase-3β
(GSK-3β), which phosphorylates β-catenin and targets it for
degradation via the ubiquitin-mediated proteasomal pathway
(57,58). The excess cytoplasmic β-catenin is
subsequently translocated into the nucleus to perform a variety of
functions such as cell proliferation, the first step in cancer
(59–61). It can act in conjunction with TCF
and LEF to regulate the expression of numerous key target genes
such as epidermal growth factor receptor (EGFR) (62). In their study, Feng et al
(17) showed that CCRK expression
in immortal liver cells induced focus formation,
anchorage-independent growth, and tumor formation in
immunodeficient mice, whereas CCRK knockdown in HCC cells reduced
G1/S phase progression, focus formation, and tumorigenicity,
demonstrating the strong oncogenic capacity of CCRK in HCC.
Moreover, β-catenin depletion reverses the CCRK-induced
tumorigenicity in nude mice, indicating that active β-catenin
signaling is a major mediator of CCRK-induced tumorigenicity. These
data are in agreement with the fact that AR silencing reduced the
expression of active β-catenin and β-catenin target genes, which
could be rescued by ectopic CCRK expression. Furthermore, the CCRK
expression correlates with AR and active β-catenin levels in
primary HCC specimens. Therefore, a vicious cycle exists: AR
induces CCRK expression to stimulate β-catenin activity, while
β-catenin, acting downstream of CCRK, induces AR expression and
activity. CCRK may therefore be a positive component in the Wnt
signaling pathway, present in the cell to aberrantly activate
β-catenin, and to provoke tumor-associated cell cycle defects to
induce unscheduled proliferation (Fig.
2).
The mechanism underlying β-catenin activation by
CCRK remains unclear. CCRK is potentially able to dysregulate
β-catenin activation. Firstly, CCRK may phosphorylate GSK3β at
Thr390. Recent studies have suggested that the C-terminal of GSK3β,
specifically phosphorylation at Thr390, is important for β-catenin
activation (63). Secondly, CCRK
may directly interact with the N-terminal domain of β-catenin,
allowing β-catenin to escape from its negative control. Thirdly,
CCRK may functionally interact with TCF, leading to the
transcription of target genes through the interaction of β-catenin
with TCF (64). However, further
investigation is needed to validate these hypotheses. Since AR and
Wnt signaling pathways are important in various types of human
cancer and CCRK overexpression was detected in a number of cancer
cells, the disruption of the AR-CCRK-β-catenin-positive regulatory
circuit is highly relevant to cancer formation in HCC, and
potentially to other male-predominant cancers.
Besides modulating the signaling pathway involved in
cell cycle control and cell proliferation, CCRK has also been shown
to regulate cyclin D1 expression. Wu et al (23) observed that CCRK overexpression in
TOV-21G (ovarian carcinoma) cells induced an upregulated expression
of cyclin D1, but not in Cdk2, pCdk2, Cdk4 and Cdk6 molecules, the
results suggesting that the mechanism by which CCRK regulates cell
cycle progression may be cell-type specific. In both the HCC and
ovarian carcinoma cell lines, CCRK overexpression caused the
upregulated expression of cyclin D1 (17,23).
Consistently, RNA interference and transfection studies have shown
that silencing of CCRK reduced the cyclin D1 expression both in
mRNA and in protein levels, and inhibited HCC and ovarian carcinoma
cell proliferation both in vitro and in vivo. In
addition, a significant correlation between the expression of CCRK
and cyclin D1 in ovarian carcinomas was observed. Studies have also
demonstrated that knockdown of β-catenin abrogated the CCRK-induced
cyclin D1 expression and G1/S phase transition of liver cells. The
overexpression of cyclin D1 was found to often associate closely
with incessant tumor growth, progression and/or poor patient
outcome. Thus, abnormally high levels of cyclin D1 are involved in
a number of GI and non-GI malignancies, including those originating
from the oral cavity, esophagus, breast and bladder (65–67).
Therefore, investigation into the molecular mechanisms and signal
transduction pathways between CCRK and cyclin D1 in various types
of cancer is crucial.
CCRK as a promising target for cancer
therapy
CCRK was overexpressed in various types of cancer
(Table I). Increasing levels of
CCRK were correlated with poor survival in patients with HCC
(17). Furthermore, a high
expression of CCRK was associated with poor prognosis in ovarian
carcinoma (23). Increased CCRK
expression affects brain tumorigenesis by upregulating CDK2
activity (21). The suppression of
CCRK by siCCRK was found to induce G1 phase cell cycle arrest and
reduced cell growth of colorectal carcinoma cells (11). Consistently, stable clones of
colorectal carcinoma cells expressing shCCRK exhibited decreased
cell proliferation rates (11).
CCRK is also involved in the human papillomavirus association with
cervical cancer (20). Moreover,
CCRK is able to regulate AR and β-catenin activity, which are
closely associated with various types of cancer (14,17).
As disruption of the cell cycle control is a hallmark of all
malignant cells, CCRK is potentially an important cell cycle
regulator during carcinogenesis. The association between CCRK and
CDK2 is well characterised at the structural and biological levels.
Several lines of evidence strongly suggest that CCRK positively
regulates cell cycle progression and tumor growth, indicating that
CCRK may serve as a novel prognostic marker and may be a promising
candidate as a molecular target for cancer therapy for some types
of cancer. The use of molecule inhibitors to block the action of
CCRK has not been previously reported.
Conclusion
Previous data have shown that CCRK is a novel CAK
and serves as an important regulator in tumorigenicity. CCRK is
functionally connected to a broad range of cell signaling pathways
with important functions in cell cycle progression, cell
proliferation and malignant transformation. The involvement of CCRK
in tumorigenesis is extensive. Numerous activities of CCRK affect
tumor growth, both in a CAK and an oncoprotein manner.
Currently, the best understood function of CCRK is
its role in phosphorylating cdk2 on Thr-160. However, several
questions concerning the function of CCRK in this pathway remain.
It is possible that siCCRK-reduced CDK2 phosphorylation is a
secondary effect of the growth inhibition induced by CCRK
knockdown. In addition, CCRK may bind and/or phosphorylate CDK7 (or
other CDKs), which in turn bind to CDK2 and regulate its activity
(21). However, more studies such
as in vitro kinase assays are needed to confirm whether CCRK
interacts directly with CDK2. In addition, previous studies suggest
that the T-loop phosphorylation of Cdk2 is completely dispensable
(68). Members of the Ringo/speedy
family proteins have now been shown to associate with Cdk2 and
promote its activity against some substrates where T-loop
phosphorylation has no effect on the Cdk2 activity (69). Future studies on the structure of
Ringo/Cdk2 complexes and more thorough genetic and biochemical
studies on Cdk2 and CCRK are required to elucidate these issues. It
is also worth investigating the relationship between CCRK and
MRK/MAK, the findings of which may reveal novel mechanisms for cell
cycle regulation.
CCRK is also capable of interacting with β-catenin
as a positive component in the Wnt signaling pathway. Future
studies are needed to elucidate the precise mechanism of action of
CCRK in regulating β-catenin. Disruption of the
AR-CCRK-β-catenin-positive regulatory circuit plays a critical role
in hepatocarcinogenesis, and may be highly relevant to the design
of therapeutic interventions in some types of cancer (17). Furthermore, the upregulated
expression of CCRK in ovarian carcinoma and HCC suggests a
potentially important role of CCRK in the control of cell
proliferation via the regulation of cyclin D1 expression (17,23).
The ultimate aim of these studies is to translate
them into clinical applications. If its role as an important
regulator in tumorigenicity is confirmed, CCRK may serve as a novel
prognostic cancer marker and a new target of cancer
therapeutics.
References
|
1.
|
DR GreenGI EvanA matter of life and
deathCancer Cell11930200210.1016/S1535-6108(02)00024-7
|
|
2.
|
Y ZwangM OrenY YardenConsistency test of
the cell cycle: roles for p53 and EGR1Cancer
Res7210511054201210.1158/0008-5472.CAN-11-338222315347
|
|
3.
|
J CicenasM ValiusThe CDK inhibitors in
cancer research and therapyJ Cancer Res Clin
Oncol13714091418201110.1007/s00432-011-1039-421877198
|
|
4.
|
J PinesFour-dimensional control of the
cell cycleNat Cell Biol1E73E79199910.1038/1104110559915
|
|
5.
|
S OrtegaM MalumbresM BarbacidCyclin
D-dependent kinases, INK4 inhibitors and cancerBiochim Biophys
Acta16027387200211960696
|
|
6.
|
K MitraC WunderB RoysamG LinJ
Lippincott-SchwartzA hyperfused mitochondrial state achieved at
G1-S regulates cyclin E buildup and entry into S phaseProc Natl
Acad Sci USA1061196011965200910.1073/pnas.090487510619617534
|
|
7.
|
D CobrinikPocket proteins and cell cycle
controlOncogene2427962809200510.1038/sj.onc.120861915838516
|
|
8.
|
G LolliLN JohnsonCAK-cyclin-dependent
activating kinase: a key kinase in cell cycle control and a target
for drugs?Cell Cycle4572577200510.4161/cc.4.4.160715876871
|
|
9.
|
A SuttonR FreimanThe Cak1p protein kinase
is required at G1/S and G2/M in the budding yeast cell
cycleGenetics147577119979286668
|
|
10.
|
MR WallenfangG Seydouxcdk-7 is required
for mRNA transcription and cell cycle progression in
Caenorhabditis elegans embryosProc Natl Acad Sci
USA9955275532200210.1073/pnas.08261839911960010
|
|
11.
|
X AnSS NgD XieFunctional characterisation
of cell cycle-related kinase (CCRK) in colorectal cancer
carcinogenesisEur J
Cancer4617521761201010.1016/j.ejca.2010.04.00720466538
|
|
12.
|
S LarochelleJ PandurRP FisherHK SalzB
SuterCdk7 is essential for mitosis and for in vivo Cdk-activating
kinase activityGenes Dev12370381199810.1101/gad.12.3.3709450931
|
|
13.
|
H CleversWnt/beta-catenin signaling in
development and
diseaseCell127469480200610.1016/j.cell.2006.10.01817081971
|
|
14.
|
BT MacDonaldK TamaiX HeWnt/beta-catenin
signaling: components, mechanisms, and diseasesDev
Cell17926200910.1016/j.devcel.2009.06.01619619488
|
|
15.
|
W YangHX YanL ChenWnt/beta-catenin
signaling contributes to activation of normal and tumorigenic liver
progenitor cellsCancer
Res6842874295200810.1158/0008-5472.CAN-07-669118519688
|
|
16.
|
Y YingQ TaoEpigenetic disruption of the
WNT/beta-catenin signaling pathway in human
cancersEpigenetics4307312200910.4161/epi.4.5.937119633433
|
|
17.
|
H FengAS ChengDP TsangCell cycle-related
kinase is a direct androgen receptor-regulated gene that drives
beta-catenin/T cell factor-dependent hepatocarcinogenesisJ Clin
Invest12131593175201110.1172/JCI4596721747169
|
|
18.
|
P KaldisMJ SolomonAnalysis of CAK
activities from human cellsEur J
Biochem26742134221200010.1046/j.1432-1327.2000.01455.x10866826
|
|
19.
|
L WohlboldS LarochelleJC LiaoG LivshitsJ
SingerKM ShokatRP FisherThe cyclin-dependent kinase (CDK) family
member PNQALRE/CCRK supports cell proliferation but has no
intrinsic CDK-activating kinase (CAK) activityCell
Cycle5546554200610.4161/cc.5.5.254116552187
|
|
20.
|
Y LiuC WuK Galaktionovp42, a novel
cyclin-dependent kinase-activating kinase in mammalian cellsJ Biol
Chem27945074514200410.1074/jbc.M30999520014597612
|
|
21.
|
SS NgYT CheungXM AnCell cycle-related
kinase: a novel candidate oncogene in human glioblastomaJ Natl
Cancer Inst99936948200710.1093/jnci/djm01117565152
|
|
22.
|
H QiuH DaiK JainR ShahC HongJ PainB TianDE
VatnerSF VatnerC DepreCharacterization of a novel cardiac isoform
of the cell cycle-related kinase that is regulated during heart
failureJ Biol
Chem2832215722165200810.1074/jbc.M71045920018508765
|
|
23.
|
GQ WuD XieGF YangYJ LiaoSJ MaiHX DengJ
SzeXY GuanYX ZengMC LinHF KungCell cycle-related kinase supports
ovarian carcinoma cell proliferation via regulation of cyclin D1
and is a predictor of outcome in patients with ovarian carcinomaInt
J Cancer12526312642200910.1002/ijc.2463019672860
|
|
24.
|
Y YangY HuHY GuN LuW LiuQ QiL ZhaoXT
WangQD YouQL GuoOroxylin A induces G2/M phase cell-cycle arrest via
inhibiting Cdk7-mediated expression of Cdc2/p34 in human gastric
carcinoma BGC-823 cellsJ Pharm
Pharmacol6014591463200810.1211/jpp.60.11.000618957166
|
|
25.
|
J YuQL GuoQD YouL ZhaoHY GuY YangHW ZhangZ
TanX WangGambogic acid-induced G2/M phase cell-cycle arrest via
disturbing CDK7-mediated phosphorylation of CDC2/p34 in human
gastric carcinoma BGC-823
cellsCarcinogenesis28632638200710.1093/carcin/bgl16817012222
|
|
26.
|
V KrystofS UldrijanCyclin-dependent kinase
inhibitors as anticancer drugsCurr Drug
Targets11291302201010.2174/13894501079071195020210754
|
|
27.
|
J Węsierska-GądekM MaurerPromotion of
apoptosis in cancer cells by selective purine-derived
pharmacological CDK inhibitors: one outcome, many mechanismsCurr
Pharm Des17256271201121348827
|
|
28.
|
JM EglyF CoinA history of TFIIH: two
decades of molecular biology on a pivotal transcription/repair
factorDNA Repair
(Amst)10714721201110.1016/j.dnarep.2011.04.02121592869
|
|
29.
|
SA PatelMC SimonFunctional analysis of the
Cdk7.cyclin H.Mat1 complex in mouse embryonic stem cells and
embryosJ Biol
Chem2851558715598201010.1074/jbc.M109.08168720231280
|
|
30.
|
K Glover-CutterS LarochelleB EricksonC
ZhangK ShokatRP FisherDL BentleyTFIIH-associated Cdk7 kinase
functions in phosphorylation of C-terminal domain Ser7 residues,
promoter-proximal pausing, and termination by RNA polymerase IIMol
Cell Biol2954555464200910.1128/MCB.00637-0919667075
|
|
31.
|
RP FisherSecrets of a double agent: CDK7
in cell-cycle control and transcriptionJ Cell
Sci11851715180200510.1242/jcs.0271816280550
|
|
32.
|
S LarochelleKA MerrickME TerretL
WohlboldNM BarbozaC ZhangKM ShokatPV JallepalliRP
FisherRequirements for Cdk7 in the assembly of Cdk1/cyclin B and
activation of Cdk2 revealed by chemical genetics in human cellsMol
Cell25839850200710.1016/j.molcel.2007.02.00317386261
|
|
33.
|
P PillaiS DesaiR PatelM SajanR FareseD
OstrovM Acevedo-DuncanA novel PKC-iota inhibitor abrogates cell
proliferation and induces apoptosis in neuroblastomaInt J Biochem
Cell Biol43784794201110.1016/j.biocel.2011.02.00221315177
|
|
34.
|
L BicaJ MeyerowitzSJ ParkerA CaragounisT
DuBM PatersonKJ BarnhamPJ CrouchAR WhitePS DonnellyCell cycle
arrest in cultured neuroblastoma cells exposed to a
bis(thiosemicarbazonato) metal
complexBiometals24117133201110.1007/s10534-010-9380-720931265
|
|
35.
|
RP FisherDO MorganA novel cyclin
associates with MO15/CDK7 to form the CDK-activating
kinaseCell78713724199410.1016/0092-8674(94)90535-58069918
|
|
36.
|
G LolliLN JohnsonRecognition of Cdk2 by
Cdk7Proteins6710481059200710.1002/prot.2137017373709
|
|
37.
|
H NagaharaSA EzhevskyAM Vocero-AkbaniP
KaldisMJ SolomonSF DowdyTransforming growth factor beta targeted
inactivation of cyclin E:cyclin-dependent kinase 2 (Cdk2) complexes
by inhibition of Cdk2 activating kinase activityProc Natl Acad Sci
USA961496114966199910.1073/pnas.96.26.1496110611320
|
|
38.
|
Z FuKA LarsonRK ChittaIdentification of
yin-yang regulators and a phosphorylation consensus for male germ
cell-associated kinase (MAK)-related kinaseMol Cell
Biol2686398654200610.1128/MCB.00816-0616954377
|
|
39.
|
RM MarshallX GranaMechanisms controlling
CDK9 activityFront Biosci1125982613200610.2741/199416720337
|
|
40.
|
WH YangJH HeatonH BrevigS MukherjeeJA
Iñiguez-LluhíGD HammerSUMOylation inhibits SF-1 activity by
reducing CDK7-mediated serine 203 phosphorylationMol Cell
Biol29613625200910.1128/MCB.00295-0819015234
|
|
41.
|
UM SachdevaJM O'BrienUnderstanding pRb:
toward the necessary development of targeted treatments for
retinoblastomaJ Clin
Invest122425434201210.1172/JCI5711422293180
|
|
42.
|
V KolupaevaC BasilicoOverexpression of
cyclin E/CDK2 complexes overcomes FGF-induced cell cycle arrest in
the presence of hypophosphorylated Rb proteinsCell
CycleJul.12012(E-pub ahead of print).
|
|
43.
|
P MeraldiJ LukasAM FryJ BartekEA
NiggCentrosome duplication in mammalian somatic cells requires E2F
and Cdk2-cyclin ANat Cell Biol18893199910.1038/1005410559879
|
|
44.
|
M StamatakosV PallaI KaraiskosK
XiromeritisI AlexiouI PaterasK KontzoglouCell cyclins: triggering
elements of cancer or not?World J Surg
Oncol8111201010.1186/1477-7819-8-11121176227
|
|
45.
|
RA WeinbergThe retinoblastoma protein and
cell cycle
controlCell81323330199510.1016/0092-8674(95)90385-27736585
|
|
46.
|
L XiaD RobinsonAH MaHC ChenF WuY QiuHJ
KungIdentification of human male germ cell-associated kinase, a
kinase transcriptionally activated by androgen in prostate cancer
cellsJ Biol Chem2773542235433200210.1074/jbc.M20394020012084720
|
|
47.
|
F DesmotsHR RussellY LeeK BoydPJ
McKinnonThe reaper-binding protein scythe modulates apoptosis and
proliferation during mammalian developmentMol Cell
Biol251032910337200510.1128/MCB.25.23.10329-10337.200516287848
|
|
48.
|
F DesmotsHR RussellD MichelPJ
McKinnonScythe regulates apoptosis-inducing factor stability during
endoplasmic reticulum stress-induced apoptosisJ Biol
Chem28332643271200810.1074/jbc.M706419200
|
|
49.
|
R MinamiM ShimadaH YokosawaH
KawaharaScythe regulates apoptosis through modulating
ubiquitin-mediated proteolysis of the Xenopus elongation
factor XEF1AOBiochem J405495501200710.1042/BJ2006188617428197
|
|
50.
|
T GoldenM SwingleRE HonkanenThe role of
serine/threonine protein phosphatase type 5 (PP5) in the regulation
of stress-induced signaling networks and cancerCancer Metastasis
Rev27169178200810.1007/s10555-008-9125-z
|
|
51.
|
BM HamH JayachandranF YangNovel Ser/Thr
protein phosphatase 5 (PP5) regulated targets during DNA damage
identified by proteomics analysisJ Proteome
Res9945953201010.1021/pr900820720039704
|
|
52.
|
AH MaL XiaSJ DesaiDL BoucherY GuanHM
ShihXB ShiRW deVere WhiteHW ChenCG TepperHJ KungMale germ
cell-associated kinase, a male-specific kinase regulated by
androgen, is a coactivator of androgen receptor in prostate cancer
cellsCancer
Res6684398447200610.1158/0008-5472.CAN-06-163616951154
|
|
53.
|
SP BalkKE KnudsenAR, the cell cycle, and
prostate cancerNucl Recept Signal6e001200818301781
|
|
54.
|
M KalraJ MayesS AssefaAK KaulR KaulRole of
sex steroid receptors in pathobiology of hepatocellular
carcinomaWorld J
Gastroenterol1459455961200810.3748/wjg.14.594518932272
|
|
55.
|
N AminE VincanThe Wnt signaling pathways
and cell adhesionFront Biosci17784804201210.2741/3957
|
|
56.
|
RS TaraporeIA SiddiquiH MukhtarModulation
of Wnt/beta-catenin signaling pathway by bioactive food
componentsCarcinogenesis33483491201210.1093/carcin/bgr30522198211
|
|
57.
|
D KimelmanW Xubeta-catenin destruction
complex: insights and questions from a structural
perspectiveOncogene2574827491200610.1038/sj.onc.121005517143292
|
|
58.
|
DM RobertsMI PronobisJS PoultonJD
WaldmannEM StephensonS HannaM PeiferDeconstructing the β-catenin
destruction complex: mechanistic roles for the tumor suppressor APC
in regulating Wnt signalingMol Biol Cell22184518632011
|
|
59.
|
A KikuchiH YamamotoA SatoS MatsumotoNew
insights into the mechanism of Wnt signaling pathway activationInt
Rev Cell Mol
Biol2912171201110.1016/B978-0-12-386035-4.00002-122017973
|
|
60.
|
CY LoganR NusseThe Wnt signaling pathway
in development and diseaseAnnu Rev Cell Dev
Biol20781810200410.1146/annurev.cellbio.20.010403.11312615473860
|
|
61.
|
S WhittakerR MaraisAX ZhuThe role of
signaling pathways in the development and treatment of
hepatocellular
carcinomaOncogene2949895005201010.1038/onc.2010.23620639898
|
|
62.
|
X TanU ApteA MicsenyiE KotsagrelosJH LuoS
RanganathanDK MongaA BellGK MichalopoulosSP MongaEpidermal growth
factor receptor: a novel target of the Wnt/beta-catenin pathway in
liverGastroenterology129285302200510.1053/j.gastro.2005.04.01316012954
|
|
63.
|
JL BuescherCJ PhielA noncatalytic domain
of glycogen synthase kinase-3 (GSK-3) is essential for activityJ
Biol Chem28579577963201010.1074/jbc.M109.09160320080974
|
|
64.
|
P Uysal-OnganerRM KyptaWnt11 in 2011 - the
regulation and function of a non-canonical WntActa Physiol
(Oxf)2045264201210.1111/j.1748-1716.2011.02297.x21447091
|
|
65.
|
V GordonS BhadelW WunderlichCDK9 regulates
AR promoter selectivity and cell growth through serine 81
phosphorylationMol
Endocrinol2422672280201010.1210/me.2010-023820980437
|
|
66.
|
JK KimJA DiehlNuclear cyclin D1: an
oncogenic driver in human cancerJ Cell
Physiol220292296200910.1002/jcp.2179119415697
|
|
67.
|
EA MusgroveCE CaldonJ BarracloughA StoneRL
SutherlandCyclin D as a therapeutic target in cancerNat Rev
Cancer11558572201110.1038/nrc309021734724
|
|
68.
|
AR NebredaCDK activation by non-cyclin
proteinsCurr Opin Cell
Biol18192198200610.1016/j.ceb.2006.01.00116488127
|
|
69.
|
T AbbasA DuttaCDK2-activating kinase
(CAK): more questions than answersCell
Cycle511231124200610.4161/cc.5.10.281916687928
|