Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of primary cancer in the world (1), ranking the sixth most prevalent cancer
and the third most frequent cause of cancer-related mortality
(2). More than half a million
individuals are diagnosed with this disease every year worldwide,
including approximately 20,000 new cases in the USA (3). Hepatocellular carcinoma has a poor
5-year survival rate of approximately 7% despite treatment
(4). Potentially curative
therapies, including liver transplantation and surgical resection,
are only applied to a minority of subjects due to the advanced
stage of the disease at the time of diagnosis and the lack of
suitable organ donors. Other regional treatments may be beneficial
for unresectable HCC, but local failure or recurrences are frequent
and the long term survival rate remains poor.
Gene therapy may offer a new therapeutic option for
HCC and is considered to be a potential adjuvant of other therapies
(5). Previous clinical trials have
shown that the side-effects are acceptable in the majority of the
cases and the mechanism of action is different from standard
treatments (6). In the past decade,
the gene therapy of liver cancer has covered a variety of gene
transfer strategies aimed to treat patients with primary and
secondary liver tumors, including gene-directed enzyme/pro-drug
therapy, inhibition of oncogenes and restoration of
tumor-suppressor genes, immunotherapy, anti-angiogenesis and
virotherapy (7). However, several
of these strategies have reached early clinical development with
little success. The main obstacles to further progress lie in the
poor gene delivery efficiency and therapeutic effect of single
genes. It is a widely accepted theory that cancer is a complex
disease, with multiple genes involved in diverse pathways.
Therefore, it may be possible to achieve a more effective control
of tumor growth through selecting effective gene delivery
approaches and an appropriate combination of therapeutic genes.
Tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL) belongs to the TNF family
(8), which functions as a cytokine
to selectively induce apoptosis in cancer cells and has minimal or
no toxicity against normal tissues in vitro and in
vivo(9,10). TRAIL is known to bind with TRAIL-R1
[death receptor (DR) 4], TRAIL-R2 (DR5), TRAIL-R3 [decoy receptor
(DcR) 1], TRAIL-R4 (DcR2) and osteoprotegerin. Among these,
TRAIL-R1 and -R2 possess intracellular death domains and,
subsequently, have the ability to mediate TRAIL-induced apoptosis
(11). Converseley, the immune
defense role of TRAIL has been shown to kill pathogen-infected or
malignant cells (12). Numerous
studies have demonstrated the potential use of recombinant soluble
TRAIL as a cancer therapeutic agent. The extracellular domain of
TRAIL works as a soluble cytokine (sTRAIL) and induces apoptosis on
cancer cells at distant locations from the producing cell (13).
Endostatin, a carboxyl-terminal proteolytic fragment
of collagen XVIII, is a key tumor suppressor and has been used
highly successfully to treat cancer. Endostatin has a multi-binding
ability to tropomyosin, heparin, perlecan, glypican, integrin,
zinc, laminin and fibulin (14). By
virtue of these binding interactions and a variety of downstream
effects, endostatin acts as a specific inhibitor of endothelial
cell proliferation, migration and angiogenesis. This results in the
inhibition of tumor growth (15,16).
Previous research has also demonstrated that endostatin may inhibit
angiogenesis in hepatocellular carcinoma following transarterial
chemoembolization (17).
Replication-defective adenoviral (Ad) vectors have
shown promise as a tool for gene delivery-based therapeutic
applications due to their high gene delivery efficiency. Moreover,
adenoviruses have a number of other advantages as gene delivery
vectors, including the ability to transduce a wide variety of
non-dividing and dividing cells with high efficiency, relative ease
of construction and ability to be purified as high-titer viral
stocks. These characteristics make adenoviruses particularly
attractive for overexpressing specific genes in vitro and
for evaluating in vivo biological activity in animal models
(18). Gendicine, the first gene
therapy product approved by China FDA, is composed of the
adenoviral vector and the human wild-type p53 tumor suppressor
gene. Clinical trials have confirmed that it is safe and effective
for head and neck squamous cell carcinoma (19).
Previous evidence has shown that carcinoma growth
and angiogenesis were suppressed by combined plasmid-mediated
endostatin and TRAIL gene therapy in mice (20). The current study reports on the use
of an adenovirus as the gene delivery vector to construct the
recombinant adenovirus Adeno-X-TRAIL (Ad-T) and Adeno-X-endostatin
(Ad-E) and to examine whether the combination of TRAIL with
endostatin works synergistically against HCC.
Materials and methods
Cell lines and animals
The human embryonic kidney HEK293 cells, human
hepatocellular carcinoma HepG2 cells and human umbilical vein
endothelial cells (HUVECs) were obtained from the American Type
Culture Collection (ATCC, Rockville, MD, USA) and maintained in
DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Life Technologies) and
antibiotics. For the HUVECs, 10 ng/ml VEGF was added to the media.
The culture was maintained in a 95% air humidified atmosphere
containing 5% CO2 at 37°C. Specific pathogen-free
six-week-old female BALB/c nude mice were obtained from Beijing
Weitong Lihua Test Animal Co. (Beijing, China). All animals were
housed under pathogen-free conditions. The animal experiments were
carried out according to the Institutional Guidelines of Tsinghua
University Graduate School at Shenzhen.
Construction of recombinant
adenovirus
Adeno-X expression system (BD Clontech, Mountain
View, CA, USA) was used to construct recombinant adenovirus
vectors. Complementary DNA encoding 184 amino acid residues of
human endostatin and 167 amino acid residues of human TRAIL (from
114 to 281 amino acids), including the signal peptide of human
interleukin-2, were PCR-amplified from pMD-18-endostatin and
pMD-18-TRAIL and subcloned into the pShuttle plasmids. The
expression cassettes, including the cytomegalovirus (CMV) promoter,
endostatin or sTRAIL gene fragments and bovine growth hormone
polyadenylation (BGH) polyA tails, were then removed from
pShuttle-endostatin and pShuttle-TRAIL by PI-SceI and
I-CeuI digestion and inserted into the corresponding PI-Sce
I and I-Ceu I sites of linearized adenoviral backbone. Thus two
recombinant adenoviral plasmids (named Ad-E and Ad-T) were
obtained. The correct recombinants were selected and retransformed
into DH5α-competent cells. Purified recombinant plasmids were
linearized by PacI restriction and transfected into HEK293
cells to generate recombinant adenoviruses. Recombinant viruses
were propagated in HEK293 cells, purified using an Adeno-X™ Maxi
purification kit and kept in a solution containing 10% glycerol, 10
mM Tris (pH 7.6) and 1 mM MgCl2. Ad-EGFP was used as a
control adenovirus expressing an enhanced green fluorescent protein
reporter gene. Viral titers were calculated by determination of the
TCID50 or with the use of the adeno-X rapid titer kit (BD
Bioscience, Palo Alto, CA, USA). Titers are expressed as either
multiplicity of infection (MOI) or as plaque-forming units
(pfu)/ml.
Western blot analysis to detect
endostatin and sTRAIL expression
HUVECs and HepG2 cells were seeded in 6-well plates
and grown to 80% confluence, at which point the culture medium was
replaced with serum-free medium and the cells were infected with
recombinant adenoviruses at an MOI of 10 or 50. The media were
collected 48 h later and the concentrated conditioned supernatants
were subjected to western blot analysis in a standard procedure.
Ad-EGFP was used for the visual examination of transgene expression
under fluorescent microscope.
Cell viability assay
Cell viability was determined by MTT assay. In
brief, 2×104 HUVECs and HepG2 cells were seeded in
96-well plates and infected with Ad-E or Ad-T, respectively, at
either 10 or 50 MOI. At 24, 48 or 72 h following virus infection,
10 μl of 5 mg/ml MTT in PBS solution was added into the media and
incubated for a further 4 h. Subsequently, the formazan product was
solubilized by addition of 100 μl of dimethyl sulfoxide. Absorbance
was measured at a wavelength of 490 nm using a microplate reader
and cellular viability (%) was determined.
Animal experiments
All animal studies were approved by our Institute’s
Animal Care and Use Committee. HepG2 cells (1×107) were
injected into the right flank of female BALB/c nude mice in 200 μl
serum-free medium at the age of 6–8 weeks. Therapy was initiated
when mice had developed palpable tumors with diameters of 5–8 mm 10
days following inoculation. Mice were randomly divided into 5
groups with 5 animals each and subjected to intratumoral injections
of virus suspension as follows: Group 1, 100 μl PBS (PBS group);
Group 2, 100 μl Ad-EGFP virus (2×109 pfu per tumor);
Group 3, 100 μl Ad-T virus (2×109 pfu per tumor); Group
4, 100 μl Ad-E virus (2×109 pfu per tumor) and Group 5,
100 μl Ad-T + Ad-E virus (1×109 pfu Ad-T and
1×109 pfu Ad-E per tumor). The treatment procedure was
repeated three more times, at 3-day intervals. Tumor growth was
monitored by caliper measurements twice a week and tumor volume was
calculated using the formula: 0.52 × (largest diameter × smallest
diameter2). The tumors were extracted for histology and
immunohistochemistry examination.
Histology and immunohistochemistry
examination
Tumor-bearing mice were sacrificed one week
following the last treatment and the tumors were dissected, covered
with Tissue-Tek (Sakura Finetek Europe B.V., Alphen aan den Rijn,
The Netherlands) and then frozen in liquid nitrogen vapor for
further histological examination. Tumor sections (5-μm thick) were
cut with a cryostat microtome (CM1950; Leica, Heidelberg, Germany)
and stained with hematoxylin and eosin (HE) as described previously
(21). Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) staining was
performed to quantitatively assess tumor apoptosis with an in
situ Cell Death Detection kit (Roche Diagnostics, Mannheim,
Germany) according to the manufacturer’s instructions. The number
of apoptotic cells was quantified by determining the percentage of
positively stained cells for all nuclei from six randomly chosen
fields/sections at ×200 magnification. Tumor sections were also
fixed in acetone, incubated and stained with an anti-CD31 antibody
to detect tumor microvessel density (MVD) as previously reported
(22). MVD was determined by
counting the number of microvessels per high-power field.
Statistical analysis
Statistical analysis was carried out with SPSS
software (version 13.0 for Windows). All values are expressed as
mean ± SD. Data were analyzed by one-way ANOVA and then differences
among the means were analyzed using the Kaplan-Meier multiple
comparison test. Differences were considered significant at
P<0.05.
Results
Expression detection of recombinant
endostatin and TRAIL
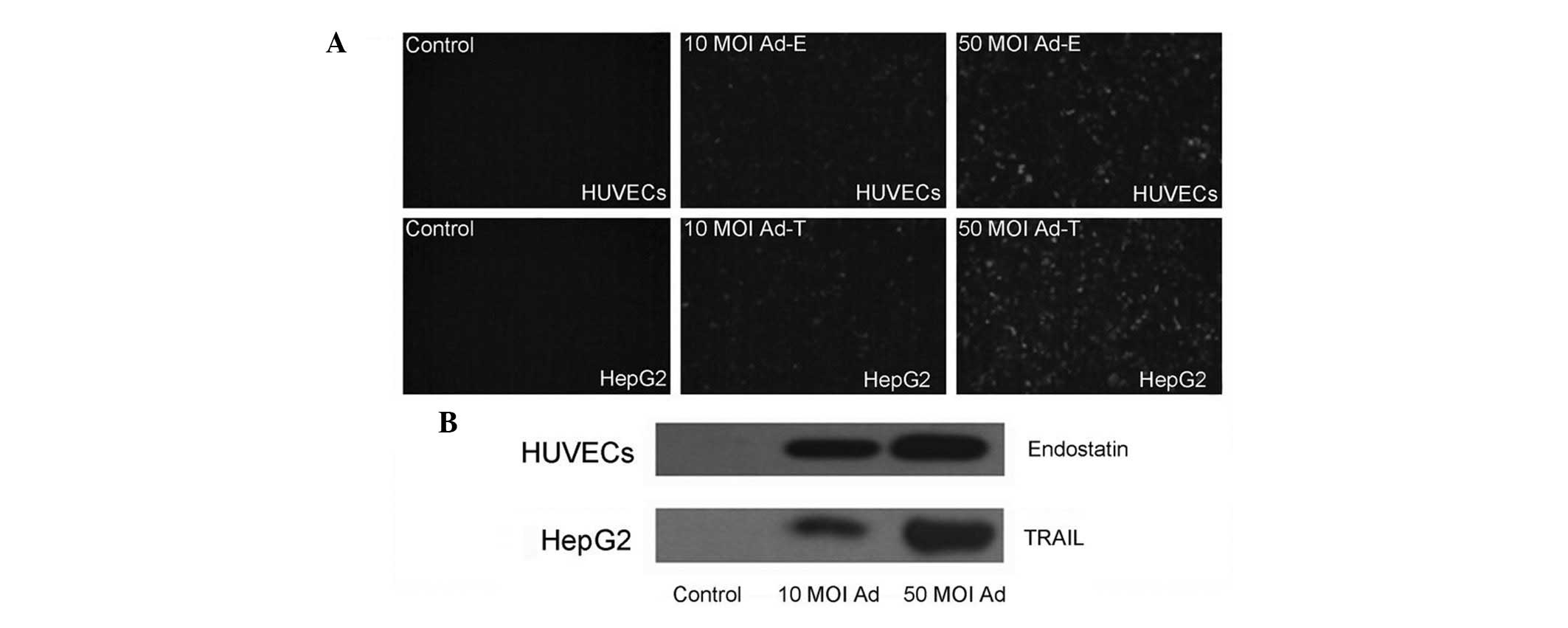
Fluorescent microscope examination confirmed the
expression of EGFP proteins in the HUVECs and HepG2 cells infected
with Ad-EGFP virus (Fig. 1A). In
order to examine whether endostatin and TRAIL genes inserted into
the adenovirus vector were able to express and secrete, the
supernatants of HUVECs and HepG2 cells infected with Ad-E or Ad-T
was detected by western blotting using anti-endostatin and
anti-TRAIL antibodies, respectively. Results revealed that an
endostatin protein band of 20 kDa and a TRAIL protein band of 18.5
kDa were able to be detected, whereas no such band was present in
the control supernatants of cells infected with Ad-EGFP virus. A
higher level expression of in 50 MOI infected cell supernatants
than that of 10 MOI infected cell supernatants following 48 h was
also observed (Fig. 1B). These
results confirmed the effective expression of transgenes by using
Adeno-X expression system.
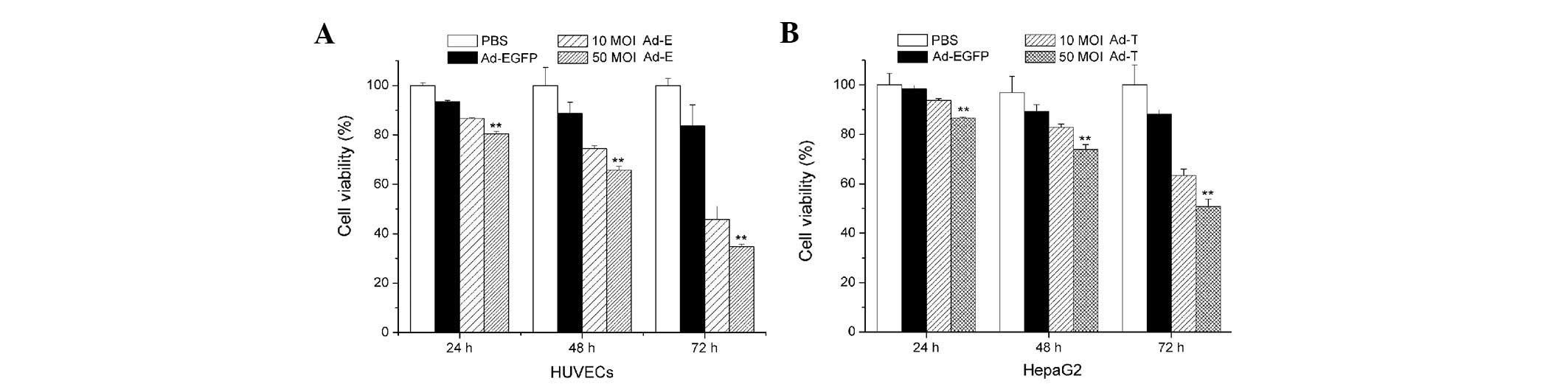
Cell viability assay
The HUVECs and HepG2 cells were infected with Ad-E
and Ad-T, respectively, using 10 or 50 MOI. Ad-EGFP adenovirus
vector served as a control. Fig. 2
shows cell growth inhibition of HUVECs and HepG2 cells treated with
Ad-E and Ad-T virus following 24, 48 or 72 h, respectively. When
HUVECs were infected with Ad-E at 10 MOI, the cell viability of
HUVECs following 24, 48 and 72 h was 93.74, 82.85 and 63.30%,
respectively. With the increasing amount of virus (50 MOI) added
into the media, a significantly lower cell viability was observed,
with 86.61, 73.92 and 50.76% following 24, 48 and 72 h virus
infection (P<0.01; Fig. 2A).
Similarly, cell growth inhibition was also observed in the
Ad-T-infected HepG2 cells. When HepG2 cells were infected with Ad-T
at 10 MOI, the cell viability of HepG2 following 24, 48 and 72 h
was 91.61, 74.48 and 45.83%, respectively. A more significant
inhibition of cell proliferation was observed when using 50 MOI
Ad-T virus infection, achieving 86.61, 73.92 and 50.76% following
24, 48 and 72 h virus infection (P<0.01; Fig. 2B). Treatment with Ad-EGFP adenovirus
vector had no significant cell growth inhibition effect on HUVECs
and HepG2 cells at various time points.
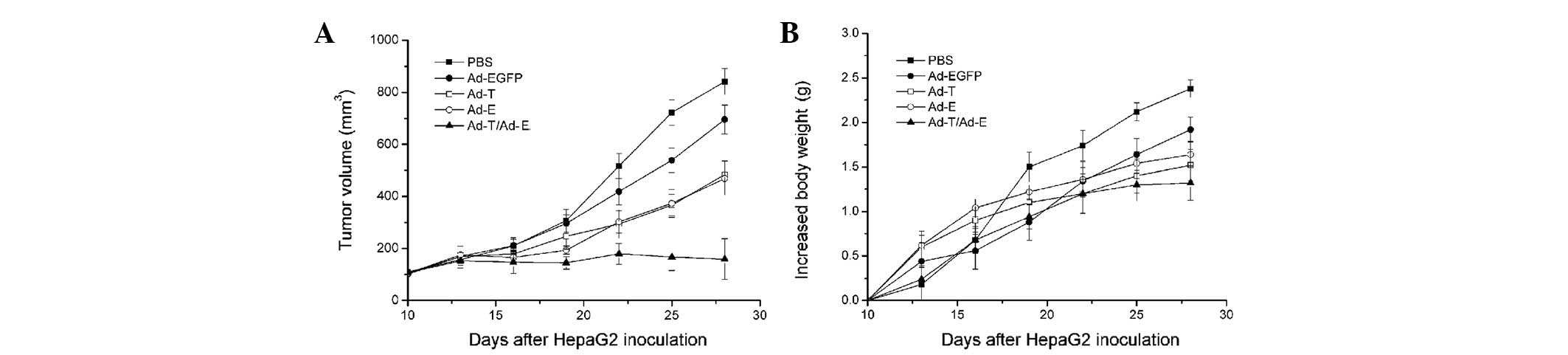
In vivo antitumor efficacy of Ad-E and
Ad-T
Subsequently, the efficacy of antitumor treatment
with Ad-E and Ad-T was investigated (Fig. 3A). In control mice treated with only
PBS, HepG2-derived tumors reached a volume of 841.47±50.32
mm3 by day 28 following implantation. There was no
significant tumor inhibition effect when mice were treated with
Ad-EGFP (695.33±55.50 mm3 tumor volume on day 28;
P>0.05). In comparison, the tumors treated with Ad-T or Ad-E
were significantly smaller than those treated with PBS control,
reaching only 483.48±52.28 and 468.76±63.73 mm3,
respectively (P<0.05). Notably, combined treatment with Ad-T +
Ad-E resulted in a significant reduction in tumor volume
(158.72±78.30 mm3), compared with all other groups on
day 28 following implantation (P<0.01). Changes in body weight
of mice demonstrated a similar trend (Fig. 3B). In the PBS-and Ad-EGFP-treated
groups, the mean increases in the body weight of mice by day 28
following implantation were 2.38±0.35 and 2.09±0.28 g,
respectively. In comparison, the mean increase in body weight of
mice treated with the single Ad-T virus was 1.64±0.24 g and that of
mice treated with the single Ad-E virus was 1.52±0.15 g. Other than
that, combined treatment with Ad-E and Ad-T virus revealed the
least increase in body weight of mice, with a 1.32±0.24 g mean
increased body weight per mouse (P<0.01).
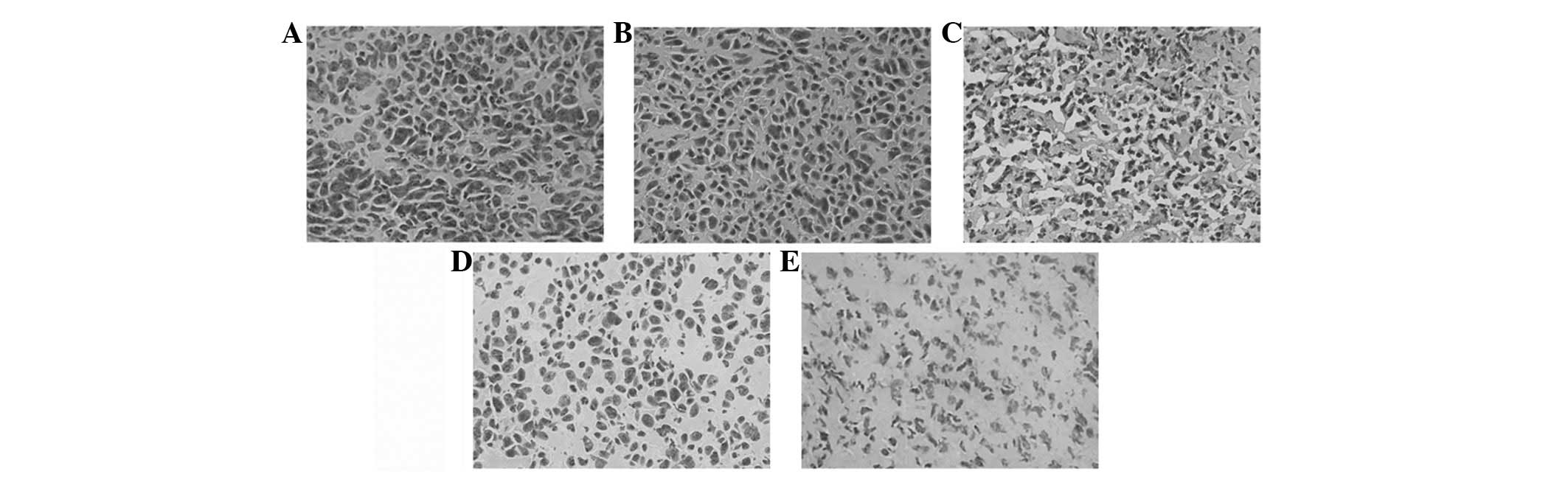
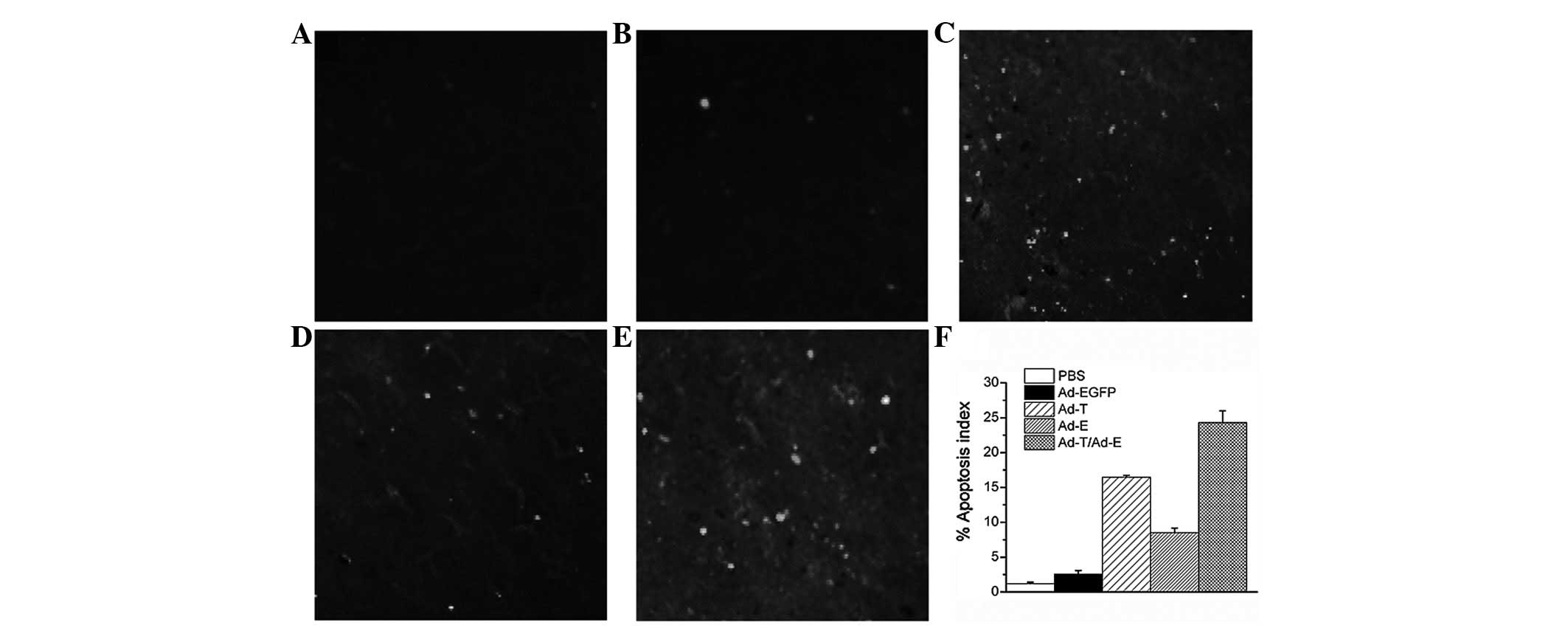
Histology and immunohistochemistry
examination
Histologically, control tumors (PBS and Ad-EGFP)
exhibited no or little tumor necrosis. Tumors treated with Ad-E or
Ad-T demonstrated a significant tumor necrosis. However, tumors
treated with Ad-E + Ad-T had the most significantly visible
homogeneous necrosis with a clearly distinguishable morphology
compared with controls (Fig. 4). A
TUNEL assay confirmed that there were extremely few apoptotic cells
in the control tumors (PBS and Ad-EGFP), with 1.18±0.21 and
2.52±0.54% apoptotic cells, respectively. Tumors treated with Ad-E
and Ad-T had 8.52 and 16.45±0.28% apoptotic cells, respectively,
but the majority of apoptotic cells were identified in the tumors
treated with Ad-E + Ad-T, achieving 24.30±1.68% apoptotic cell
ratio (Fig. 5; P<0.01).
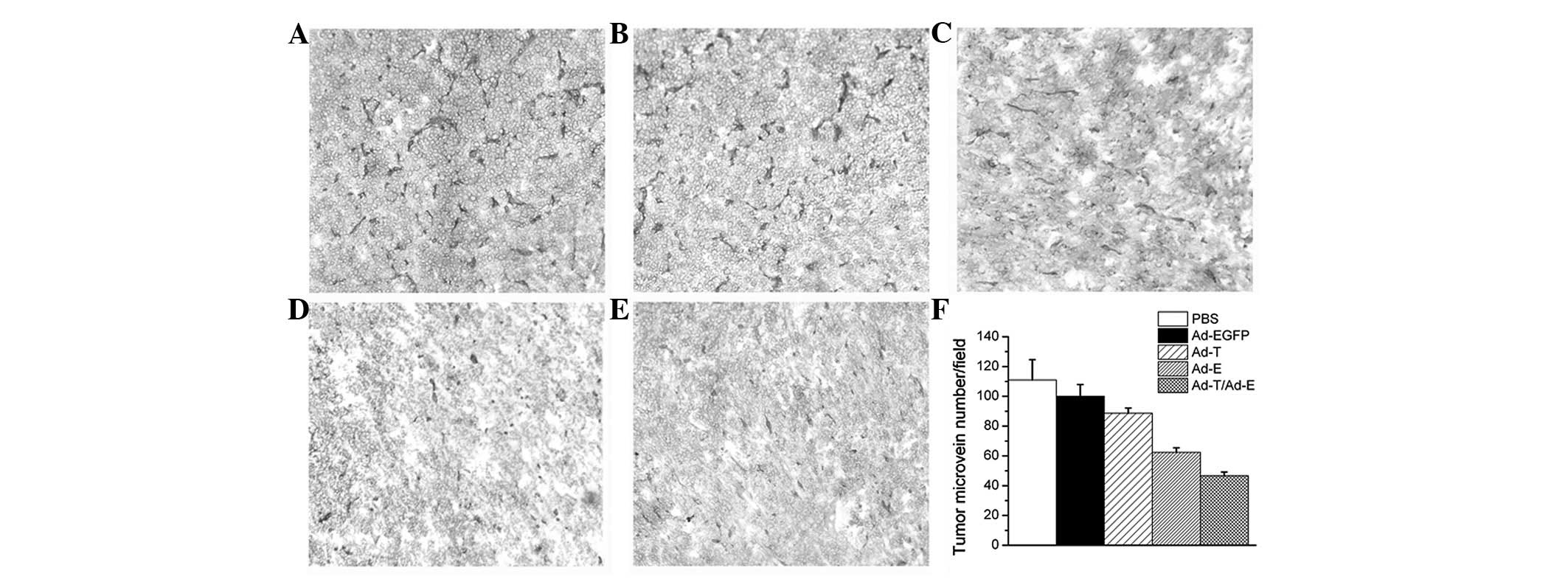
Angiogenesis in tumor tissues was estimated by MVD assay. As shown
in Fig. 6, treatment with Ad-E +
Ad-T resulted in a significant reduction in tumor MVD compared with
the PBS-, Ad-EGFP-, Ad-E or Ad-T virus-treated groups (P<0.01).
Quantitation revealed 46.76±2.51 tumor microvessels per field in
the tumors treated with Ad-E + Ad-T, which was significantly lower
compared with the PBS (111.24±13.60 microvessels), Ad-EGFP
(98.06±8.24 microvessels), Ad-T (88.67±3.52 microvessels) and Ad-E
group (62.34±3.06 microvessels).
Discussion
HCC, similar to other malignant tumors, is a complex
disease with multiple genes in diverse pathways involved in its
initiation, progression, invasion and metastasis. It is widely
accepted that the sequential accumulation of mutations that
activate oncogenes and disrupt tumour suppressor genes, combined
with multiple cycles of clonal selection and evolution, facilitate
the process of carcinogenesis. Moreover, during the growth of a
tumor, cancer metastasis may occur by virtue of a series of
aggressive steps, including leaving the primary mass,
intravasation, survival in circulation, extravasation and
colonization and growth of tumor cells at a distant site. The
promotion of tumor cell apoptosis and inhibition of angiogenesis
provides a good chance of preventing cancer from overgrowing and
becoming malignant (23,24). TRAIL is regarded as one of the most
potent inhibitors of tumor growth. Anti-angiogenic factors,
including endostatin, have been identified and have demonstrated
the ability to inhibit tumor growth in vivo(25). Therefore, a combination gene therapy
of TRAIL and endostatin is likely to achieve an improved control of
tumor growth through the combination of TRAIL-induced tumor cell
apoptosis and endostatin-induced anti-angiogenesis, as demonstrated
in the presented study (Fig. 3).
The efficacy of enhancement of tumor treatment has been
demonstrated by the combination of TRAIL or endostatin with
chemotherapy and radiotherapy (26–29).
The key point of gene therapy is to establish an
effective gene delivering system. Our data indicate that the
combination gene therapy of adenovirus-mediated TRAIL with
endostatin exhibits significant antitumor activities through
induction of apoptosis and inhibition of angiogenesis, compared
with single Ad-T or Ad-E. Our results are in agreement with the
previous study by Zhang et al(20), in which sTRAIL and endostatin genes
were transferred using plasmid pVAX1 as a vector. By contrast, the
Adeno-X expression system was utilized in our study. The Adeno-X
expression system offers a simple yet powerful method for
constructing recombinant adenovirus by using a ligation-based
method. The Adeno-X expression system is also considered as the
most efficient adenoviral system available and generates high-level
protein expression in a wide variety of mammalian host cells.
Furthermore, viral vectors have been preferentially applied in gene
therapy due to their marked advantages over the current
plasmid-transfection methods, including i) the protocols are simple
to perform, involving the direct addition of virus to cells; ii)
the use of Ad-gene vectors does not require any additional
reagents; and iii) observable adenoviral transduction efficiency is
high and reproducible in a number of the cell types tested when
used at an optimal MOI.
The current study revealed that the ratio of
apoptotic cells significantly increased in the tumors treated with
a combination of Ad-E and Ad-T virus, in comparison with signal
gene treatment (Fig. 5). The
enhanced cytotoxic sensitivity of tumor cells may be attributed to
the following several factors. The overexpression of endostatin may
result in the inhibition of angiogenesis and deprive cancer cells
of the nutritional supplements that they require for sustaining
their high metabolic activity. Thus, nutrient deficiency reprograms
cancer cells to enter into apoptosis. Previous evidence has shown
that nutrition deprivation causes cancer cell death (30). Braun et al demonstrated that
serum-nutrient starvation induces cell death mediated by Bax and
Puma (31). However, under the
condition of nutrient/oxygen depletion, cancer cells are expected
to be more sensitive towards an apoptotic inducer. Although there
are reports that the majority of HCC cells are insensitive towards
TRAIL-induced apoptosis, it is likely that HCC cells tend to be
more sensitive to TRAIL-induced apoptosis when tumors are subjected
to hypoxic and nutrient-depleted environments. In addition, it was
also notable that intratumoral MVD significantly decreased in the
residual tumors with combination therapy, compared with Ad-E- or
Ad-T-treated tumors (Fig. 6),
suggesting that a stronger in vivo anti-angiogenic effect is
obtained when Ad-E is coadministered with Ad-T. Thus, a synergistic
antitumor effect against xenograft growth of HCC is achieved
through the combined administration of Ad-E and Ad-T.
In the current study, the recombinant Ad-E and Ad-T
were sucessfully constructed by means of the adeno-X expression
system. Intratumoral administration revealed that a combination
treatment employing one-half the dose of Ad-E and Ad-T led to a
significant enhanced regression of the tumors compared with
treatment with either agent alone. Treated xenografts by
combination of Ad-E with Ad-T demonstrated increased apoptosis and
reduced angiogenesis in the tumors that may account for the
histological observation of tumor growth inhibition. Our findings
highlight a promising application prospect in achieving more
effective growth inhibition of HCC by means of adenovirus-mediated
combinatorial gene therapy.
Acknowledgements
The authors are grateful for the
financial support from the National Natural Science Foundation of
China (Grant No. 30900749), the Doctorate Fund of National
Education Ministry of China (Grant No. 20090450416) and the
Shenzhen Municipal Government and Bureau of Science, Technology and
Information through the programs of Shenzhen National Key Lab of
Health Science and Technology and the Key Lab of Gene and Antibody
Therapy.
References
|
1
|
Cha C, DeMatteo RP and Blumgart LH:
Surgery and ablative therapy for hepatocellular carcinoma. J Clin
Gastroenterol. 35(Suppl 2): S130–S137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar
|
|
3
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carr BI: Hepatocellular carcinoma: current
management and future trends. Gastroenterology. 127:S218–S224.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Di Maio M, De Maio E, Perrone F, Pignata S
and Daniele B: Hepatocellular carcinoma; systemic treatments. J
Clin Gastroenterol. 35(Suppl 2): S109–S114. 2002.PubMed/NCBI
|
|
6
|
Prieto J, Qian C, Hernandez-Alcoceba R,
Gonzalez-Aseguinolaza G, Mazzolini G, Sangro B and Kramer MG: Gene
therapy of liver diseases. Expert Opin Biol Ther. 4:1073–1091.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hernández-Alcoceba R, Sangro B and Prieto
J: Gene therapy of liver cancer. Ann Hepatol. 6:5–14. 2007.
|
|
8
|
Cosman D: A family of ligands for TNF
receptor superfamily. Stem Cells. 12:440–445. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashkenazi A and Dixit VM: Death receptors:
signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Walczak H, Miller RE, Ariail K, et al:
Tumoricidal activity of tumor necrosis factor-related
apoptosis-inducing ligand in vivo. Nat Med. 5:157–163. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Piras V, Hayashi K, Tomita M and
Selvarajoo K: Enhancing apoptosis in TRAIL-resistant cancer cells
using fundamental response rules. Sci Rep. 1:1442011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang A, Wilson NS and Ashkenazi A:
Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical
translation. Curr Opin Cell Biol. 22:837–844. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nagane M, Huang HJ and Cavenee WK: The
potential of TRAIL for cancer chemotherapy. Apoptosis. 6:191–197.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamaguchi N: An analysis of the functional
mechanisms of endostatin-the anti-angiogenic activity of endostatin
is mediated by its multiple binding ability. Connect Tissue.
36:171–178. 2004.
|
|
15
|
O’Reilly MS, Boehm T, Shing Y, et al:
Endostatin: an endogenous inhibitor of angiogenesis and tumor
growth. Cell. 88:277–285. 1997.
|
|
16
|
Yamaguchi N, Anand-Apte B, Lee M, et al:
Endostatin inhibits VEGF-induced endothelial cell migration and
tumor growth independently of zinc binding. EMBO J. 18:4414–4423.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bao Y, Feng WM, Tang CW, Zheng YY, Gong HB
and Hou EG: Endostatin inhibits angiogenesis in hepatocellular
carcinoma after transarterial chemoembolization.
Hepatogastroenterology. 59:1566–1568. 2012.PubMed/NCBI
|
|
18
|
Franceschi RT and Ge C: Gene delivery by
adenoviruses. Methods Mol Biol. 455:137–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee J and Moon C: Current status of
experimental therapeutics for head and neck cancer. Exp Biol Med.
236:375–389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y, Qu ZH, Cui M, Guo C, Zhang XM, Ma
CH and Sun WS: Combined endostatin and TRAIL gene transfer
suppresses human hepatocellular carcinoma growth and angiogenesis
in nude mice. Cancer Biol Ther. 8:466–473. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei YQ, Wang QR, Zhao X, et al:
Immunotherapy of tumors with xenogeneic endothelial cells as a
vaccine. Nat Med. 6:1160–1166. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wei YQ, Huang MJ, Yang L, et al:
Immunogene therapy of tumors with vaccine based on Xenopus
homologous vascular endothelial growth factor as a model antigen.
Proc Natl Acad Sci USA. 98:11545–11550. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ingber D, Fujita T, Kishimoto S, Sudo K,
Kanamaru T, Brem H and Folkman J: Synthetic analogues of fumagillin
that inhibit angiogenesis and suppress tumor growth. Nature.
348:555–557. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
O’Reilly MS, Holmgren L, Shing Y, et al:
Angiostatin: a novel angiogenesis inhibitor that mediates the
suppression of metastases by a Lewis lung carcinoma. Cell.
79:315–328. 1994.PubMed/NCBI
|
|
25
|
Sauter BV, Martinet O, Zhang WJ, Mandeli J
and Woo SL: Adenovirus-mediated gene transfer of endostatin in vivo
results in high level of transgene expression and inhibition of
tumor growth and metastases. Proc Natl Acad Sci USA. 97:4802–4807.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin T, Zhang L, Davis J, et al:
Combination of TRAIL gene therapy and chemotherapy enhances
antitumor and antimetastasis effects in chemosensitive and
chemoresistant breast cancers. Mol Ther. 8:441–448. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wissink EH, Verbrugge I, Vink SR, et al:
TRAIL enhances efficacy of radiotherapy in a p53 mutant, Bcl-2
overexpressing lymphoid malignancy. Radiother Oncol. 80:214–222.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu DS, Wu CM, Huang TH and Xie QD:
Combined effects of radiotherapy and endostatin gene therapy in
melanoma tumor model. Radiat Environ Biophys. 47:285–291. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu LP, Xing J, Wang QX, et al:
Therapeutic efficacy of recombinant human endostatin combined with
chemotherapeutics in mice-transplanted tumors. Eur J Pharmacol.
617:23–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim Y: The effects of nutrient depleted
microenvironments and delta-like 1 homologue (DLK1) on apoptosis in
neuroblastoma. Nutr Res Pract. 4:455–461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Braun F, Bertin-Ciftci J, Gallouet AS,
Millour J and Juin P: Serum-nutrient starvation induces cell death
mediated by Bax and Puma that is counteracted by p21 and unmasked
by Bcl-xL inhibition. PLoS One. 6:e235772011. View Article : Google Scholar : PubMed/NCBI
|