Introduction
Osteosarcoma (OS) is the most common primary bone
malignancy, comprising ∼35% of all types of bone cancer. This
disease is the eighth most common type of cancer among children; it
accounts for 2.4% of all pediatric malignancies (1). For patients with OS, the use of
chemotherapy with surgical resection alone has improved survival
from 11% in the 1960s, to 70% by the mid-1980s (2). However, survival has since plateaued,
regardless of advances in anticancer therapy (2). Elucidation of the mechanisms of
chemoresistance and implementation of strategies to overcome
chemoresistance will likely be pivotal to improving survival in OS
patients.
TWIST, also known as TWIST1, belongs to the basic
helix-loop-helix (bHLH) transcription factor family. During
embryonic development, TWIST is essential in the specification of
the mesoderm and the differentiation of mesoderm-derived tissues
(3). TWIST haploinsufficiency has
been demonstrated to impair bone development in both mice and
humans (4,5). A high expression of TWIST has been
detected in several types of cancer and has been associated with
the initial phase of metastatic progression (6). By contrast, in a homogeneous cohort of
OS patients (7), the TWIST
gene was frequently deleted in the tumors at diagnosis, and its
haploinsufficiency was significantly correlated with a poorer
patient outcome (3).
Endothelin-1 (ET-1), which promotes tumor cell
proliferation and survival through the endothelin A receptor
(ETAR), is expressed in a range of malignancies (8). Both ET-1 and ETAR are expressed in OS
cells and tissue (9,10). Felx et al demonstrated that
ET-1 is important in OS metastasis; it may promote OS cell invasion
by inducing the synthesis of MMP-2 through ETAR (9). Zhao et al revealed that
increased ET-1 expression was associated with an increased
malignancy of OS, and that ET-1 promoted OS cell invasion and
survival against cisplatin-induced apoptosis through ETAR,
suggesting that ET-1/ETAR signaling may be a potential therapeutic
target for OS metastasis as well as a target for overcoming OS cell
chemoresistance (9,10).
We have performed the first study that investigates
the interaction between TWIST and ET-1/ETAR signaling in OS cells,
and we have analyzed how this functional interaction may affect OS
cell survival against chemotherapy agent-induced apoptosis.
Materials and methods
Cells lines, plasmids and reagents
The human OS cell lines, Saos-2 and MG-63, were
purchased from the American Type Culture Collection (Rockville, MD,
USA). Human TWIST cDNA was subcloned into a pcDNA 3.1 expression
vector as described previously by Matsuo et al(11). The following were purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA): TWIST
(sc-38604-V) short hairpin RNA (shRNA) lentiviral particles,
control shRNA lentiviral particles-A (sc-108080), anti-TWIST
(sc-81417) antibody, anti-ET-1 (sc-21625) antibody, anti-Akt
(ser473) (sc-24500) antibody and anti-P-Akt (ser473) (sc-101629)
antibody. All secondary antibodies were purchased from Jackson
ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). The ET-1
enzyme-linked immunosorbent assay (ELISA) kit was purchased from
R&D Systems (Minneapolis, MN, USA), while the DeadEnd™
Fluorometric terminal deoxynucleotidyl transferase mediated
nick-end labeling (TUNEL) system was purchased from Promega
(Madison, WI, USA) and the Superfect™ transfection reagent was
purchased from Qiagen (Valencia, CA, USA). Puromycin, cisplatin,
synthetic ET-1, LY294002, BQ123 and all chemicals of reagent grade
were purchased from Sigma (St. Louis, MO, USA).
The study was approved by the ethics committee of
Xiangya Hospital, Central South University, Changsha, China.
Transfection and lentiviral
transduction
The Superfect transfection reagent (Qiagen) was used
to transfect Saos-2 cells according to the manufacturer’s
instructions. Selection with puromycin (5 μg/ml) was then
employed to generate pools of stable transductants according to the
manufacturer’s instructions. The TWIST-shRNA lentiviral
particles contained expression constructs that encoded
target-specific 19–25 nt (plus hairpin) shRNA designed to
specifically knockdown TWIST gene expression; while the
control shRNA lentiviral particles contained a scrambled shRNA
sequence that was not able to inititate the degradation of any
cellular mRNA, and was used as a negative control for TWIST
shRNA lentiviral particles. Subsequently, Saos-2 and MG-63 cells
were transduced with the lentiviral particles. Selection with
puromycin (5 μg/ml) was then used to generate pools of
stable transductants according to the manufacturer’s instructions
(Santa Cruz Biotechnology, Inc.).
Real-time quantitative reverse
transcription PCR
TRIzol reagent followed by purification with the
TURBO DNA-free system (Ambion; Austin, TX, USA) was used to prepare
RNA from brain tissue samples. The cDNA was synthesized using the
SuperScript II reverse transcriptase (Invitrogen Life Technologies,
Inc.; Carlsbad, CA, USA). Using the SYBR-Green I kit (Roche) as
described by the manufacturer, real-time quantitative PCR was
performed in the LightCycler thermal cycler system (Roche
Diagnostics; Indianapolis, IN, USA). The results were normalized
against those of the housekeeping gene glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) in the same sample. The following
primer sequences were used: Forward: 5′-TCCTCTGCTGGTTCCTGACT-3′ and
reverse: 5′-CAGAAACTCCACCCCTGTGT-3′ for human ET-1; forward:
5′-GACTCATGACCACAGTCCATGC-3′ and reverse:
5′-AGAGGCAGGGATGATGTTCTG-3′ for human GAPDH. Each experiment
was repeated twice and performed in triplicate.
Western blot analysis
The ET-1 ELISA kit was used to assess the secreted
levels of ET-1 in the cell culture supernatants. In brief, cells
were grown to confluence in 10-cm dishes in Roswell Park Memorial
Institute (RPMI)-1640 medium supplemented with 10% fetal bovine
serum (FBS), which was then replaced with serum-free medium. The
cells were incubated for a further 16 h, and then cell culture
supernatants were collected for ELISA according to the
manufacturer’s instructions (R&D Systems). ELISA-detected ET-1
concentrations are presented as the fold change relative to that of
the normal control cells (designated as 1) and were normalized
against cell number (per 106 cells). Each ELISA
experiment was repeated three times and performed in duplicate. In
the western blot analyses, protein was extracted using a lysis
buffer comprising 150 mM NaCl; 2% Triton; 0.1% sodium dodecyl
sulfate (SDS); 50 mM Tris, pH 8.0 and 10% protease inhibitor
cocktail (Sigma), and stored at -20°C. Equal amounts of protein (25
μg) for each sample were loaded onto pre-cast 7.5% Mini
Protean TGX gels (Bio-Rad, Hercules, CA, USA). Prior to loading
onto the gels, the proteins were quantified and equal loading was
verified by Ponceau coloration. The proteins were separated by
electrophoresis for 50 min at 200 V, then transferred onto a
polyvinylidene fluoride (PVDF) transfer membrane (Amersham
Biosciences/GE Healthcare; Piscataway, NJ, USA) for 55 min at 100
V. Membranes were incubated with a 1/500 dilution of anti-TWIST,
anti-MMP-2 or anti-ET-1 antibody for 1 h. Following washing,
secondary antibodies with horseradish peroxidase conjugate (1/5000;
1 h) were used to reveal the membranes. A GE Healthcare enhanced
chemiluminescence (ECL) kit was used to reveal the peroxidase
activity.
Measurement of apoptosis by TUNEL
assay
The TUNEL assay was performed using the DeadEnd
Fluorometric TUNEL system according to the instructions provided by
Promega. Cells were treated with cisplatin (10 nM) in the presence
or absence of ET-1 (10 or 100 pM) and/or LY294002 (50 μM) or
BQ123 (5 μM) for ≤8 h. A standard fluorescein filter was
used to detect the green nuclear fluorescence emitted by apoptotic
cells. A blue nuclear fluorescence was emitted by all cells stained
with 4′,6-diamidino-2-phenylindole (DAPI). The relative number of
apoptotic cells was determined by counting the number of
TUNEL-positive cells in five random fields in each sample, by
fluorescence microscopy of the slides (magnification, ×100).
Statistical analyses
Statistical analyses were performed using the
Statistical Package for the Social Sciences (SPSS)for Windows,
version 10.0 (SPSS, Inc.; Chicago, IL, USA). Data values were
expressed as mean ± standard deviation. A one-way analysis of
variance (ANOVA) followed by post hoc pairwise comparisons using
the least significant difference method were used to compare means
between multiple groups. Two-tailed α=0.05 was used as the
significance level for this study.
Results
TWIST is expressed at a very low level in Saos-2
cells (12), but is widely
detectable in MG-63 cells (13).
Saos-2 cells were stably transfected with a TWIST expression vector
to overex-press TWIST, and MG-63 cells were stably transduced with
TWIST-shRNA to knock down TWIST expression, in order to study the
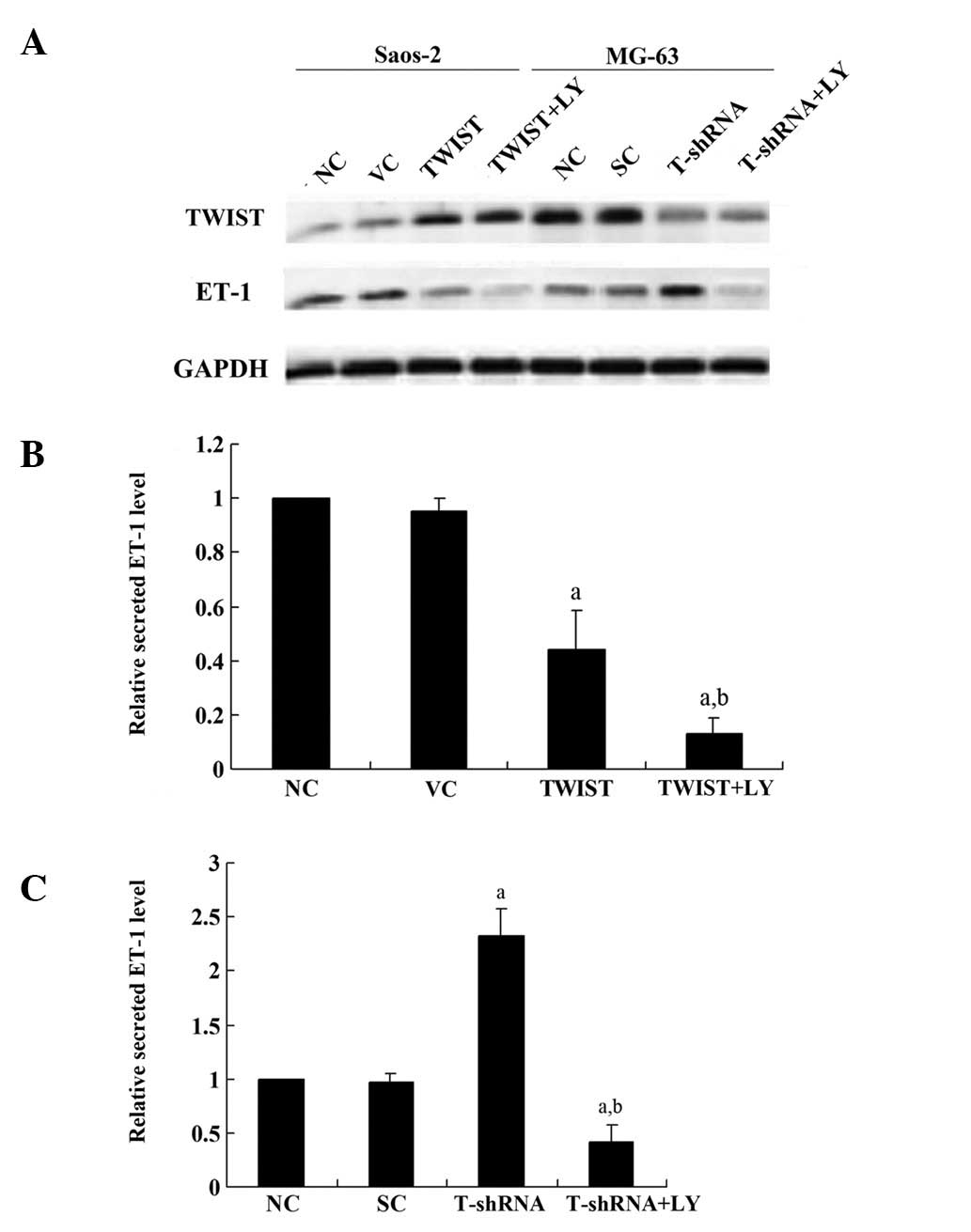
interaction between TWIST and ET-1 in OS cells. Fig. 1 demonstrates that TWIST was
overexpressed >3-fold in the Saos-2 cells, and the endogenous
level of TWIST was decreased by >75% in MG-63 cells, compared
with the control cells. Additionally, ET-1 exhibited a higher
constitutive level in Saos-2 cells than in MG-63 cells.
Overexpression of TWIST decreased the ET-1 level ∼2-fold in Saos-2
cells compared with the controls, which was significantly
strengthened by the selective phosphatidylinositol 3-kinase (PI3K)
inhibitor LY294002. Knocking down TWIST increased the ET-1 level
>2-fold in MG-63 cellscompared with the controls, which was
abolished by LY294002. Similar secreted ET-1 level results were
demonstrated in both cell lines, which suggests that TWIST inhibits
ET-1 expression in a PI3K-dependent manner in OS cells.
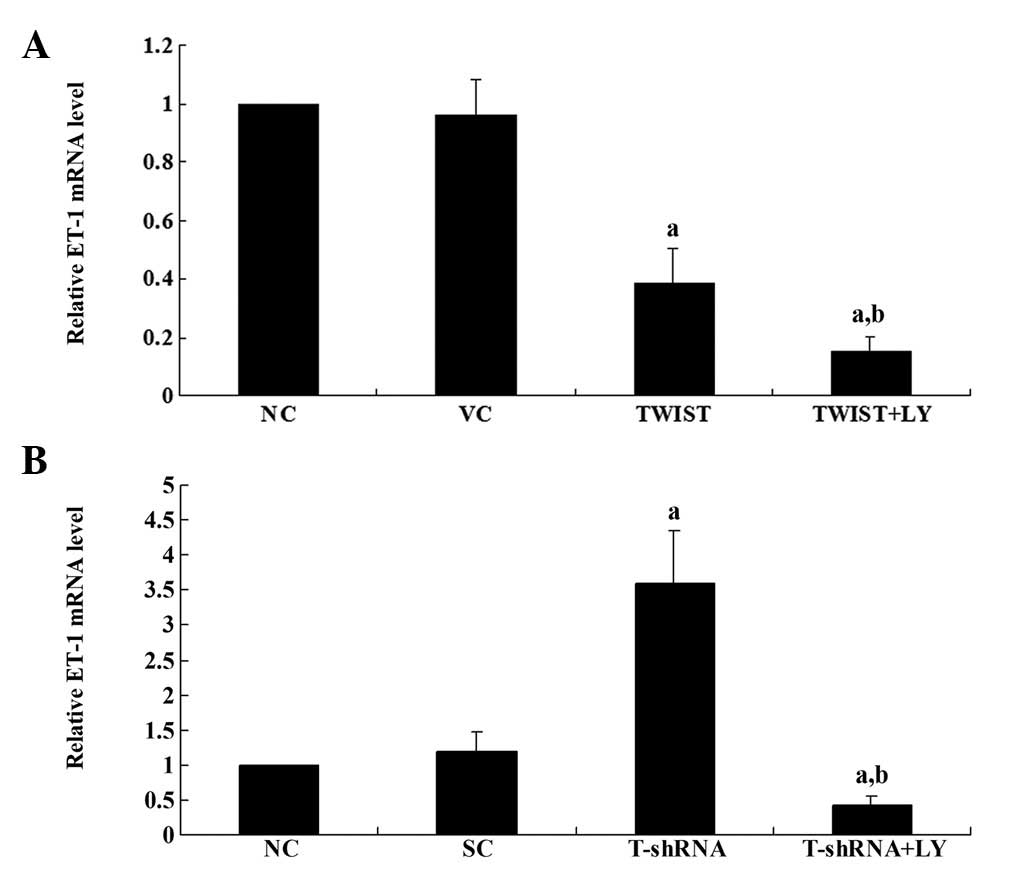
Real-time RT-PCR results showed that in Saos-2
cells, overexpression of TWIST decreased the ET-1 mRNA level
>2-fold compared with the controls, which was strengthened by
LY294002 (Fig. 2A). Additionally,
in MG-6 cells, knockdown of TWIST increased the ET-1 mRNA level
>3-fold compared with the controls, which was abolished by
LY294002 (Fig. 2B). Therefore,
TWIST inhibits the expression of ET-1 at the transcriptional level
in a PI3K-dependent manner in OS cells.
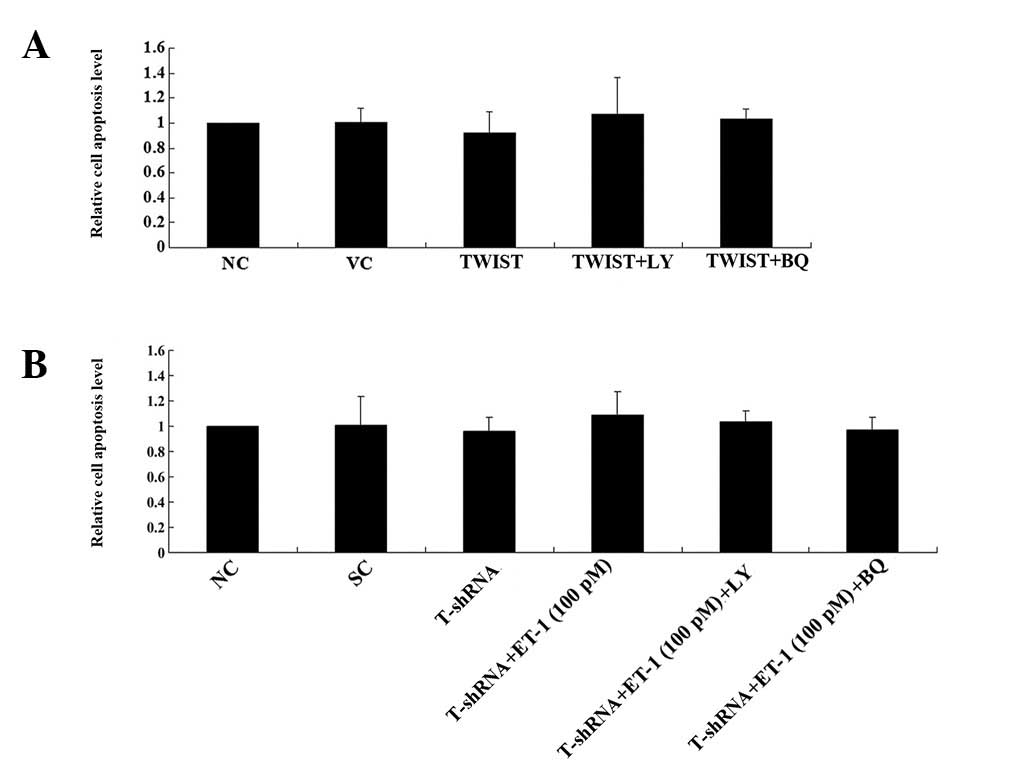
Subsequently, to investigate the effect of the
interaction between TWIST and ET-1/ETAR signaling on OS survival,
the cell apoptosis rate in both cell lines treated with 10 nM of
cisplatin was examined. Cisplatin is an apoptosis-inducing
chemotherapeutic agent typically used in OS treatment. Under normal
culture conditions, overexpressing or knocking down TWIST in the
presence or absence of ET-1 (100 pM) and/or LY294002 (50 μM)
or BQ123 (5 μM) for ≤8 h was not observed to signifcantly
affect the rate of cell apoptosis (Fig.
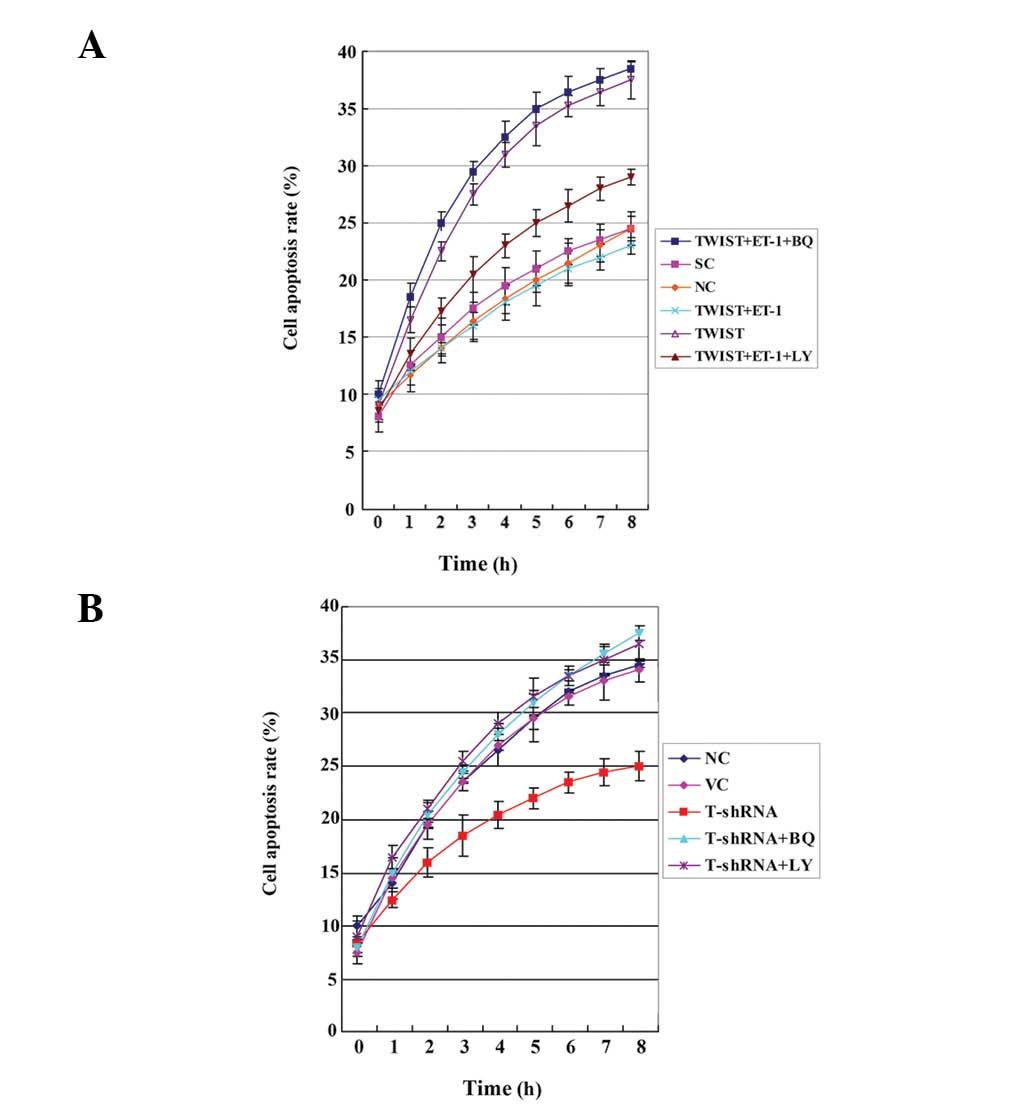
3). However, in Saos-2 cells treated with cisplatin,
overexpressing TWIST led to a significantly increased cell
apoptosis rate compared with the controls, which was reversed by
exogenous ET-1 (100 pM). Additionally, the selective ETAR inhibitor
(BQ123) was able to completely block the rescue effect of ET-1,
while LY294002 partially blocked the rescue effect (Fig. 4A). In MG-63 cells treated with
cisplatin, knocking down TWIST significantly decreased the cell
apoptosis rate. This effect was was abolished by BQ123 or LY294002
(Fig. 4B).
The results suggest that TWIST significantly
decreases OS cell survival against cisplatin by inhibiting
ET-1/ETAR signaling, which functions downstream of PI3K. The
PI3K/Akt pathway has been demonstrated to activate ET-1 gene
transcription (14), in accordance
with our finding that the regulatory effects of TWIST on ET-1
expression and ET-1/ETAR signaling may be completely blocked by
LY294002. Thus, we subsequently tested the effect of overexpressing
or knocking down TWIST on the PI3K/Akt survival signaling pathway.
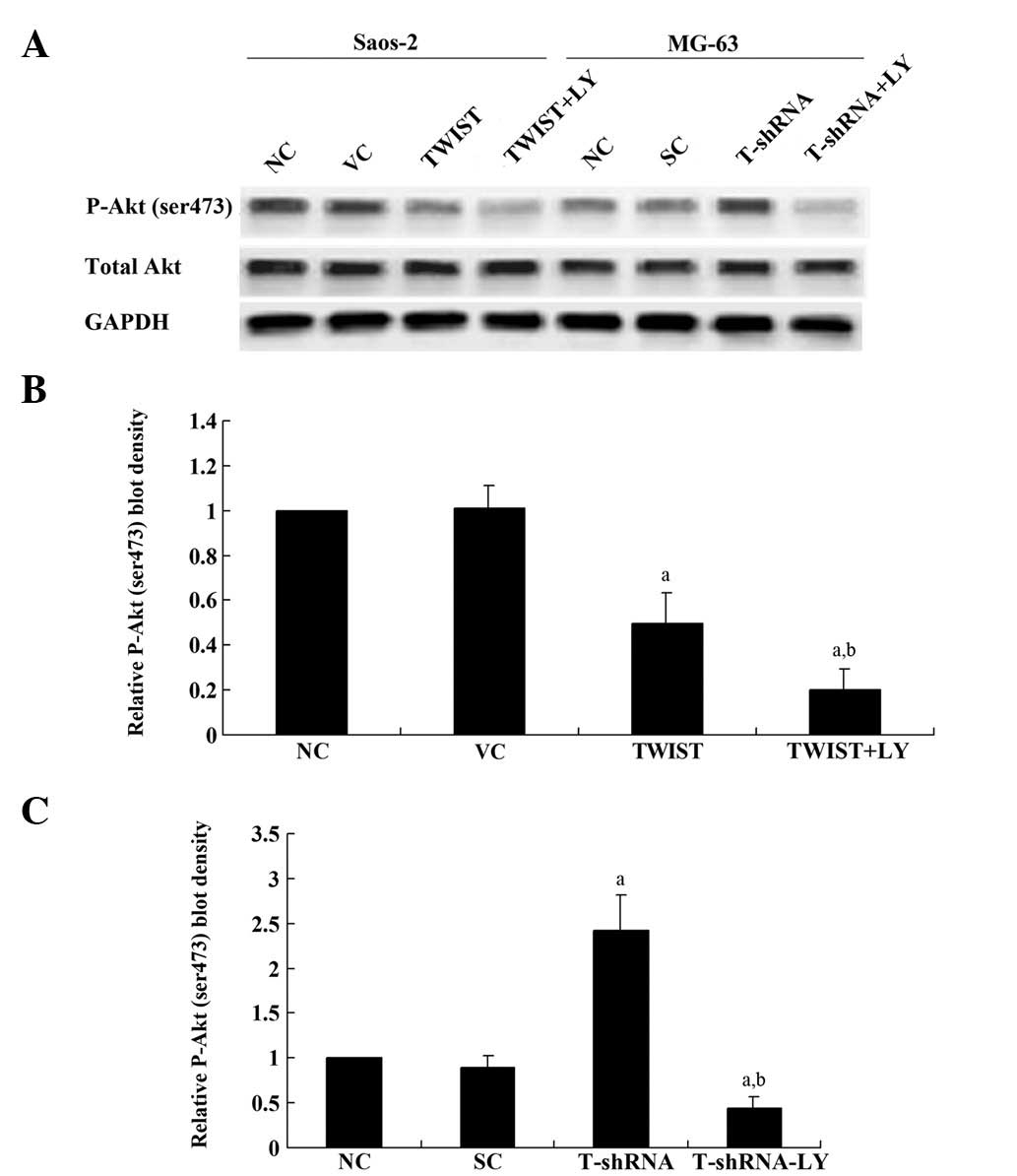
In Saos-2 cells, overexpression of TWIST significantly decreased
phosphorylation at serine 473 (ser473) of Akt, which is required
for complete activation of Akt; LY294002 treatment further
decreased phosphorylation at this site (Fig. 5A and B). In MG-63 cells, knockdown
of TWIST increased the phosphorylation of Akt (P-Akt) at ser473
≥2-fold compared with the controls, which was abolished by LY294003
(Fig. 5A and C). Overall, our
results suggest that TWIST downregulates ET-1/ETAR signaling by
inhibiting the PI3K/Akt pathway.
Discussion
The ET-1/ETAR signaling pathway is considered to be
a potential therapeutic target in the control of OS metastasis and
chemoresistance (9,10). Although a high expression of TWIST
has been described in several types of cancer and is associated
with the initial phase of metastatic progression, the TWIST
gene is frequently deleted in OS at diagnosis, and its
haploinsufficiency is significantly correlated with a poorer
patient outcome (6). In the present
study, to our knowledge, we have conducted the first investigation
into the functional interaction between TWIST and ET-1/ETAR
signaling in OS cells. We assessed the effect of this interaction
on OS cell survival against chemotherapy agent-induced
apoptosis.
Saos-2 cells express a low level of TWIST and have
been used as a model to investigate TWIST function (12). By contrast, TWIST is amply expressed
in MG-63 cells (13). Therefore,
overexpression and knockdown of TWIST were performed in the two
cell lines, respectively, to approach the study objectives. The
results of our study demonstrated that Saos-2 cells expressed a
relatively higher constitutive level of ET-1 compared with MG-63
cells, suggesting that the constitutive levels of TWIST and ET-1 in
OS cells may be negatively correlated. Further studies with
additional OS cell lines are required to address the issue. In
accordance with these findings, knockdown of TWIST in MG-63 cells
increased the ET-1 expression level as well as cell survival
against cisplatin, which was reversed by a PI3K inhibitor. Our
results were concordant with those of previous studies; they
demonstrated that TWIST regulates ET-1 expression at the
transcriptional level in a PI3K-dependent manner in OS cells.
Guenou et al demonstrated that TWIST haploinsufficiency
results in PI3K accumulation and activation of PI3K/Akt-dependent
osteoblast growth (14), while Kim
et al revealed that activated PI3K was required for the
activation of AP-1 and subsequent ET-1 gene transcription (15). In the present study, to our
knowledge, we have provided the first evidence that TWIST
downregulates ET-1 expression by inhibiting the PI3K/Akt pathway in
OS cells.
Both TWIST and ET-1/ETAR signaling reportedly
promote tumor cell survival and chemoresistance in human cancer
(16–18). However, our results suggest that
TWIST decreases cell survival against chemotherapy agents by
down-regulating ET-1/ETAR signaling, which is in accordance with
the study demonstrating that TWIST haploinsufficiency was
significantly correlated with a poorer patient outcome (3). ET-1 itself is an activator of the
PI3K/Akt pathway (19,20), while TWIST inhibits it, which
explains why exogenous ET-1 was able to completely restore cell
survival against cisplatin in OS cells overexpressing TWIST.
However, exogenous ET-1 was only partially able to rescue cell
survival in OS cells overexpressing TWIST in the presence of an
extremely high concentration of LY294002, implicating the
involvement of other signaling pathways downstream of PI3K/Akt
(i.e., other than ET-1/ETAR signaling) in the inhibitory effects of
TWIST on OS cell survival against chemotherapy agents.
Cisplatin elicits DNA repair mechanisms by
cross-linking DNA, and this activates apoptosis when repair is not
possible (21). It is not yet
understood whether the functional interaction between TWIST and
ET-1 is able to affect OS cell survival against additional types of
chemotherapy agents. Further studies with further types of
chemotherapy agents and OS cell lines would elaborate this
issue.
In conclusion, we demonstrate that TWIST
significantly decreases OS cell survival against cisplatin by
downregulating ET-1/ETAR signaling via inhibition of the PI3K/Akt
pathway. To our knowledge, our study provides the first evidence of
a functional interaction between TWIST and ET-1/ETAR signaling in
OS cells, providing novel insights into the molecular mechanisms
that underlie OS progression, cell survival and
chemoresistance.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2010. View Article : Google Scholar
|
|
2
|
Chou AJ and Gorlick R: Chemotherapy
resistance in osteosarcoma: current challenges and future
directions. Expert Rev Anticancer Ther. 6:1075–1085. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Entz-Werlé N, Lavaux T, Metzger N, et al:
Involvement of MET/TWIST/APC combination or the potential role of
ossification factors in pediatric high-grade osteosarcoma
oncogenesis. Neoplasia. 9:678–688. 2007.PubMed/NCBI
|
|
4
|
Stoetzel C, Weber B, Bourgeois P,
Bolcato-Bellemin AL and Perrin-Schmitt F: Dorso-ventral and
rostro-caudal sequential expression of M-twist in the
postimplantation murine embryo. Mech Dev. 51:251–263. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
el Ghouzzi V, Le Merrer M, Perrin-Schmitt
F, et al: Mutations of the TWIST gene in the Saethre-Chotzen
syndrome. Nat Genet. 15:42–46. 1997.
|
|
6
|
Entz-Werlé N, Choquet P, Neuville A, et
al: Targeted apc;twist double-mutant mice: a new model of
spontaneous osteosarcoma that mimics the human disease. Transl
Oncol. 3:344–353. 2010.PubMed/NCBI
|
|
7
|
Le Deley MC, Guinebretière J, Gentet JC,
et al: SFOP OS94: a randomised trial comparing preoperative
high-dose methotrexate plus doxorubicin to high-dose methotrexate
plus etoposide and ifosfamide in osteosarcoma patients. Eur J
Cancer. 43:752–761. 2007.PubMed/NCBI
|
|
8
|
Nelson J, Bagnato A, Battistini B and
Nisen P: The endothelin axis: emerging role in cancer. Nat Rev
Cancer. 3:110–116. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Felx M, Guyot MC, Felx M, Guyot MC, Isler
M, Turcotte RE, Doyon J, Khatib AM, Leclerc S, Moreau A and
Moldovan F: Endothelin-1 (ET-1) promotes MMP-2 and MMP-9 induction
involving the transcription factor NF-kappaB in human osteosarcoma.
Clin Sci (Lond). 110:645–654. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao Y, Liao Q, Zhu Y and Long H:
Endothelin-1 promotes osteosarcoma cell invasion and survival
against cisplatin-induced apoptosis. Clin Orthop Relat Res.
469:3190–3199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsuo N, Shiraha H, Fujikawa T, et al:
Twist expression promotes migration and invasion in hepatocellular
carcinoma. BMC Cancer. 9:2402009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okamura H, Yoshida K and Haneji T:
Negative regulation of TIMP1 is mediated by transcription factor
TWIST1. Int J Oncol. 35:181–186. 2009.PubMed/NCBI
|
|
13
|
Funato N, Ohtani K, Ohyama K, Kuroda T and
Nakamura M: Common regulation of growth arrest and differentiation
of osteo-blasts by helix-loop-helix factors. Mol Cell Biol.
21:7416–7428. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guenou H, Kaabeche K, Dufour C, Miraoui H
and Marie PJ: Down-regulation of ubiquitin ligase Cbl induced by
twist haploinsufficiency in Saethre-Chotzen syndrome results in
increased PI3K/Akt signaling and osteoblast proliferation. Am J
Pathol. 169:1303–1311. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim YJ, Koo TY, Yang WS, Han NJ, Jeong JU,
Lee SK and Park SK: Activation of spleen tyrosine kinase is
required for TNF-α-induced endothelin-1 upregulation in human
aortic endothelial cells. FEBS Lett. 586:818–826. 2012.
|
|
16
|
Wang X, Ling MT, Guan XY, Tsao SW, Cheung
HW, Lee DT and Wong YC: Identification of a novel function of
TWIST, a bHLH protein, in the development of acquired taxol
resistance in human cancer cells. Oncogene. 23:474–482. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun P, Xiong H, Kim TH, Ren B and Zhang Z:
Positive inter-regulation between β-catenin/T cell factor-4
signaling and endothelin-1 signaling potentiates proliferation and
survival of prostate cancer cells. Mol Pharmacol. 69:520–531.
2006.
|
|
18
|
Zhao Y, Liao Q, Zhu Y and Long H:
Endothelin-1 promotes osteosarcoma cell invasion and survival
against cisplatin-induced apoptosis. Clin Orthop Relat Res.
469:3190–3199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu MH, Lo JF, Kuo CH, et al: Endothelin-1
promotes MMP-13 production and migration in human chondrosarcoma
cells through FAK/PI3K/Akt/mTOR pathways. J Cell Physiol.
227:3016–3026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kulasekaran P, Scavone CA, Rogers DS,
Arenberg DA, Thannickal VJ and Horowitz JC: Endothelin-1 and
transforming growth factor-beta1 independently induce fibroblast
resistance to apoptosis via AKT activation. Am J Respir Cell Mol
Biol. 41:484–493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosenberg B, Vancamp L, Trosko JE and
Mansour VH: Platinum Compounds: a new class of potent antitumour
agents. Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|