Introduction
Colorectal cancer remains one of the most prevalent
health problems worldwide and it is the third most common type of
cancer and the second most common cause of cancer-related mortality
(1). The use of cytotoxic agents,
such as cisplatin (CDDP), is an effective and important treatment
for colon cancer (2,3). However, the efficacy of these drugs is
limited by the effects of chemoresistance, which is associated with
AKT overexpression (4). Thus,
methods to decrease the expression of AKT have received much
attention.
AKT/PKB is a serine/threonine kinase and belongs to
a family of proteins which includes AKT1, AKT2 and AKT3. The AKT
pathway regulates diverse cellular processes, including cell
proliferation, differentiation, apoptosis and tumorigenesis
(5,6). In addition, hyperactivation of AKT has
been detected in colon cancers that have acquired CDDP resistance
(7). The activation of AKT induces
cell survival, while the inhibition of AKT activity increases the
rate of apoptosis in numerous types of cancer cells (8). The activation of AKT has been detected
in human tumors with acquired chemoresistance (9–11).
These observations highlight AKT as an emerging target for
overcoming chemoresistance in colon cancer.
Cytoplasmic Janus protein tyrosine kinases (JAKs)
regulate multiple signaling pathways that govern cell
proliferation, differentiation and apoptosis (12). The JAK kinases regulate members of
the signal transducers and activators of transcription (STAT)
family (13). Once STAT is
tyrosine-phosphorylated by JAKs, it dimerizes and translocates to
the nucleus to activate the expression of genes, such as AKT. The
JAK2-STAT3 signaling pathway is activated by reactive oxygen
species (ROS) and this pathway activation is inhibited by
antioxidants. CDDP generates ROS and may activate the JAK2/STAT3
pathway (14). Upon phosphorylation
of the tyrosine residues by JAKs, STAT3 is activated to upregulate
AKT expression. The AKT pathway has been extensively investigated.
However, the transcriptional regulation of AKT remains largely
unknown. In the present study, a sequence from the human AKT
promoter, which may contain the binding sites for STAT3, was
selected. AKT is upregulated by STAT3 at the transcriptional level
and the overexpression of AKT is diminished by ROS inhibition. The
study demonstrated that AKT activation was closely associated with
chemoresistance in human tumors. The present results also showed
that the JAK2/STAT3 pathway mediates AKT expression, which
represents a novel target for overcoming CDDP resistance in human
tumors.
Material and methods
Cell culture and reagents
HCT-116 colon cancer cells were obtained from the
Cell Bank of the Chinese Academy of Sciences (Shanghai, China),
supplemented with 10% fetal bovine serum (FBS), 100 mg/l penicillin
and 100 mg/l streptomycin and maintained at 37°C in Dulbecco’s
modified Eagle’s medium (DMEM) in a humidified atmosphere of 5%
CO2. The following reagents were used: anti-AKT,
anti-STAT3, anti-phosphoJAK2 and anti-phosphoSTAT3, anti-phosphoAKT
(Millipore, Billerica, MA, USA), anti-JAK2 (Cell Signaling
Technology, Inc., Danvers, MA, USA), JAK2 small interfering
(si)RNA, control siRNA (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA), dichlorodihydrofluorescein diacetate (DCFDA) and
N-acetylcysteine (NAC), (Sigma, St. Louis, MO, USA).
Measurement of ROS production by flow
cytometry
HCT-116 cells were treated with CDDP for 6 h. After
washing with PBS, the cells were incubated with 33 mg/ml DCFDA
(Sigma) in PBS for 30 min at 37°C. The excess probe was washed off
with PBS and the labeled cells were measured using flow cytometric
analysis.
JAK2 siRNA transfection
Briefly, colon cells were transfected with JAK2
siRNA and control siRNA according to the manufacturer’s
instructions. Fresh medium without antibiotics was added after 8 h
of incubation and after an additional 48 h, the medium was replaced
with 10% fetal bovine serum (FBS), 100 mg/l penicillin and 100 mg/l
streptomycin and cell growth was maintained.
Isolation of RNA and quantitative
RT-PCR
The mRNA levels of AKT in the various CDDP-treated
cells were analyzed by quantitative RT-PCR. Total RNA was extracted
using TRIzol (Invitrogen, Carlsbad, CA, USA), according to the
manufacturer’s instructions. The total cDNA was then used in the
PCR to measure the mRNA levels of AKT. The mRNA level of actin was
used as the internal control. The conditions were as follows:
pre-denaturing at 94°C for 4 min and 30 cycles of 94°C for 30 sec,
56°C for 30 sec and 72°C for 25 sec. The sequence of the upstream
primer used for AKT was 5′-TCT ATG GCG CTG AGA TTG TG-3′ and the
downstream primer sequence was 5′-CTT AAT GTG CCC GTC CTT GT-3′.
For actin, the upstream primer was 5′-CAC GAT GGA GGG GCC GGA CTC
ATC-3′ and the downstream primer was 5′-TAA AGA CCT CTA TGC CAA CAC
AGT-3′.
Western blotting
The cells were solubilized in ice-cold lysis buffer
(1X PBS, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS and
10 mg/ml phenyl). Following 30 min of centrifugation at 13,000 × g,
4°C, the supernatants were transferred to new microcentrifuge tubes
and the protein concentration of the supernatant was measured using
the BCA protein assay (Pierce Biotechnology, Inc., Rockford, IL,
USA) and subsequently stored at −80°C. Cell lysate (50 μg)
was separated on an SDS-polyacrylamide gel. Following SDS-PAGE, the
proteins were transferred to nitrocellulose membranes. To detect
the proteins, the membranes were blocked using 10% non-fat dry milk
in Tris buffer containing 0.1% Tween-20 (TBS-T) and then incubated
at 4°C overnight with anti-AKT and anti-phosphoJAK2,
anti-phosphoSTAT3, anti-phosphoAKT antibodies, which were diluted
in TBS-T containing 5% non-fat dry milk.
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed as described previously
(15). Solubilized chromatin was
prepared from a total of 1×107 HCT-116 cells treated
with CDDP and NAC (an inhibitor of ROS). The chromatin solution was
diluted 10-fold with ChIP dilution buffer (SDS lysis buffer and
protease inhibitor cocktail), then added to Protein G Agarose and
rotated at 4°C for 1 h. The pre-cleared chromatin solution was
divided and utilized in the immunoprecipitation assays with either
an anti-Stat3 antibody or an anti-actin antibody. After washing
with buffers (low/high salt immune complex, TE buffer), the
antibody-protein-DNA complex was eluted from the beads by
resuspending the pellets in 20% SDS, 1 M NaHCO3 and
H2O at room temperature for 15 min. After cross-linking,
the protein and RNA were removed by incubating the sample with 1
μl RNase A at 37°C for 30 min and then cross-linking with
the pellets in 0.5 M EDTA, 8 μl 1 M Tris-Hcl and 1 μl
proteinase K at 45°C for 2 h. Purified DNA was subjected to PCR
with primers specific to the putative Stat3-binding site within the
AKT promoter. The conditions were as follows: pre-denaturing at
94°C for 4 min and 30 cycles of 94°C for 30 sec, 65°C for 30 sec
and 72°C for 25 sec. The sequences of the PCR primers used were as
follows: forward 5′-CTT CGT GAA CAT TAA CGA CAG GGC C-3′; reverse,
5′-AAT GGC CAC CCT GAC TAA GGA GTG G-3′.
Statistical analysis
Differences were analyzed with the χ2
test for incidence data and the Student’s t-test for comparisons of
the means. P<0.05 was considered to indicate statistically
significant differences.
Results
CDDP-induced activation of AKT in HCT-116
cells
DNA-damaging reagents, such as CDDP, are known to
trigger cancer cell death. However, the efficacy of these agents is
limited due to chemoresistance, which is associated with AKT
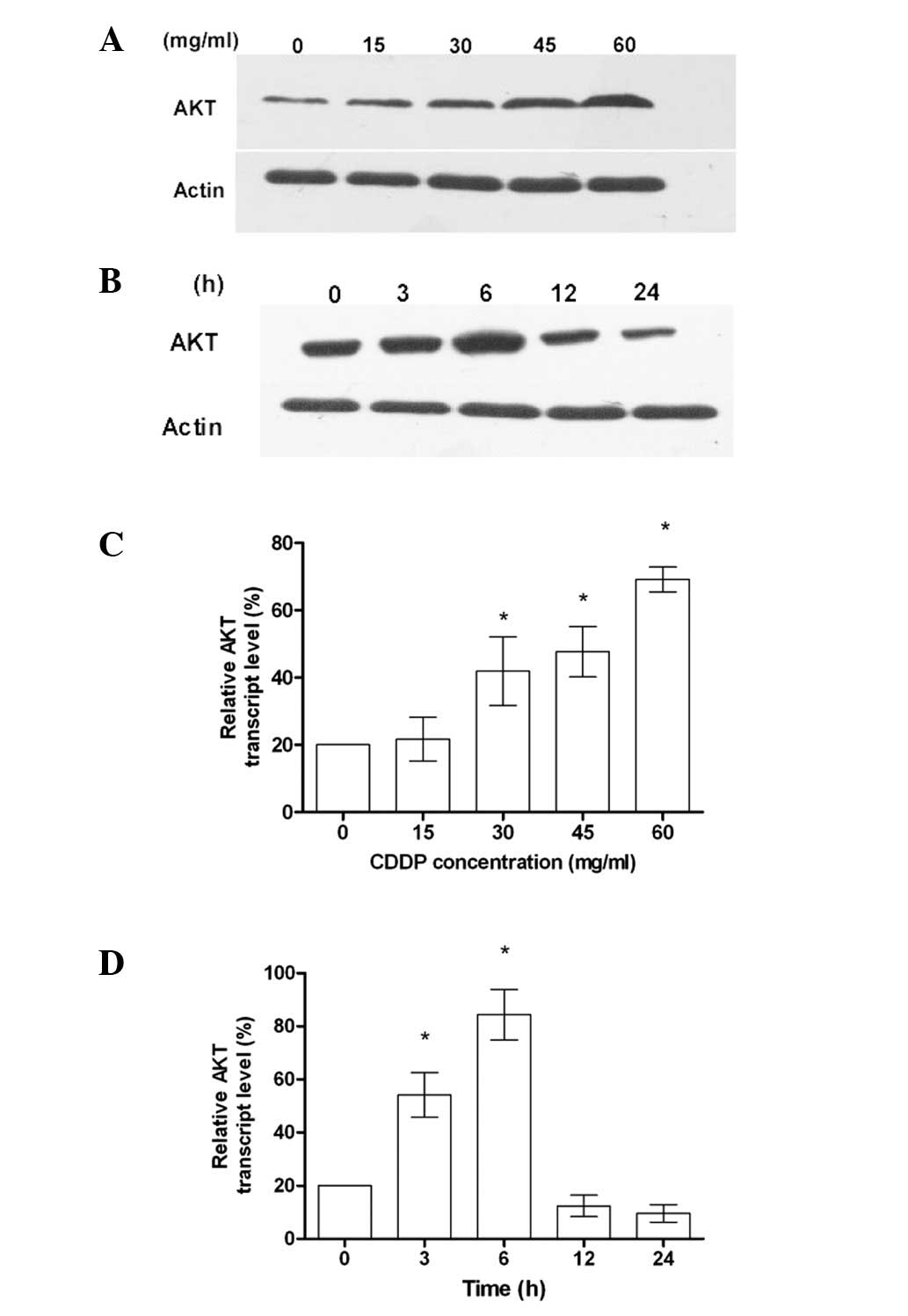
overexpression. As shown in Fig. 1A and
C, the increased expression of AKT was dependent on the
concentration of CDDP. When the cells were treated with CDDP at a
constant concentration, the level of AKT expression at 6 h was
significantly increased (Fig. 1B and
D). These results suggest that AKT gene overexpression is a
mechanism for cells exhibiting chemotherapy drug resistance in
human colon cancer. Treatment of HCT-116 cells with CDDP induced
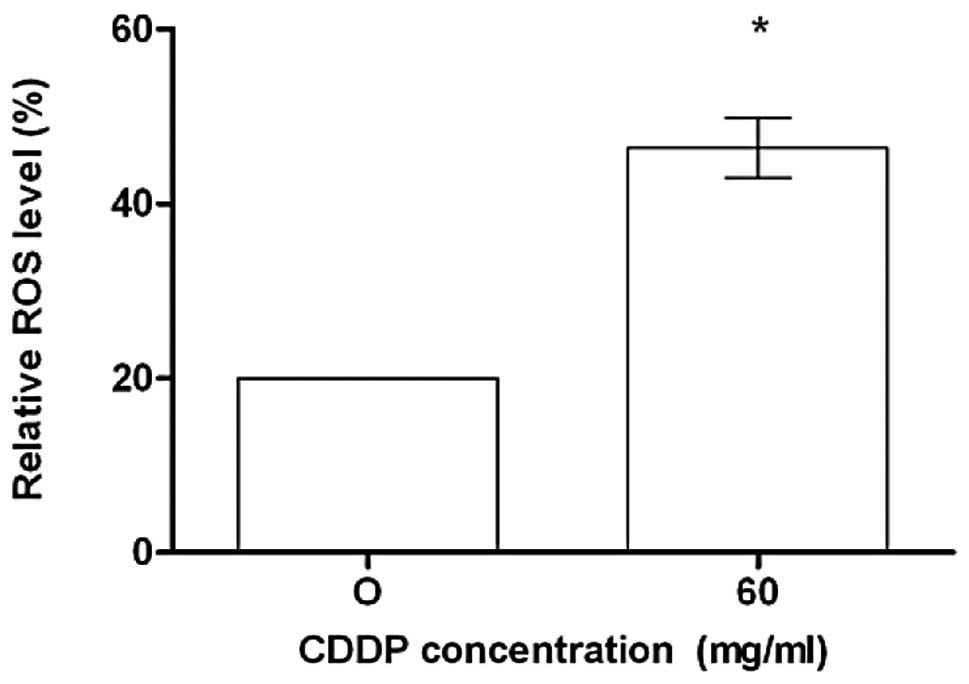
the generation of hydrogen peroxide and superoxide anions, as
measured by the oxidation of the DCFDA. As shown in Fig. 2, constitutive ROS were observed in
CDDP-treated cells.
To test the hypothesis that the JAK/STAT signaling
pathway activation is associated with ROS in HCT-116 cells,
growth-arrested HCT-116 cells were treated with 60 mg/ml CDDP or 33
mg/ml NAC at concentrations that were able to significantly affect
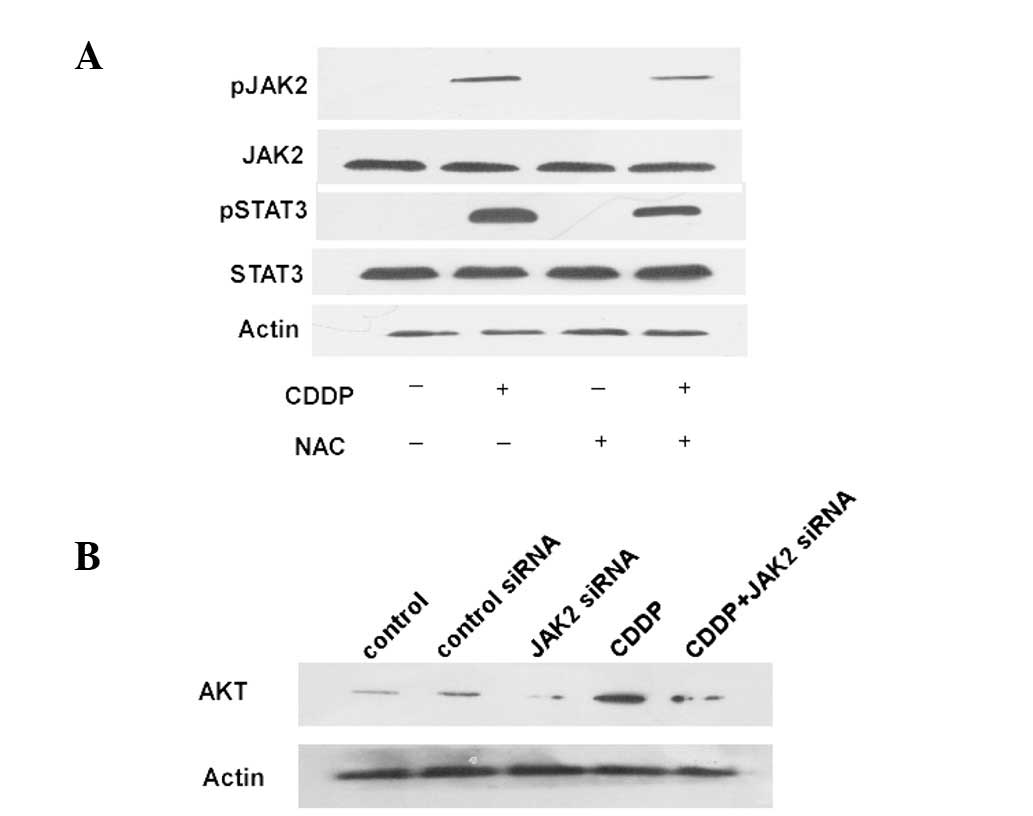
AKT activation. CDDP caused rapid tyrosine phosphorylation of JAK2
(Fig. 3A). As shown in Fig. 3A, constitutive tyrosine
phosphorylation of STAT3 was observed in the CDDP-treated cells.
Reducing the expression of JAK2 in colon cancer cells using JAK2
siRNA decreases the AKT expression levels (Fig. 3B). These experiments demonstrate the
phosphorylation and nuclear translocation of STAT3 in
CDDP-stimulated HCT-116 cells. Taken together with the results
shown in Fig. 3, these findings
indicate that CDDP activates the JAK2/STAT3 pathway through the
generation of ROS in HCT-116 cells.
STAT3 increases AKT expression
To analyze the transcriptional regulation of the AKT
gene, a sequence from the human AKT promoter gene was selected. To
demonstrate whether STAT3 binds directly to the STAT3-binding site
within the AKT promoter and that AKT is transcriptionally regulated
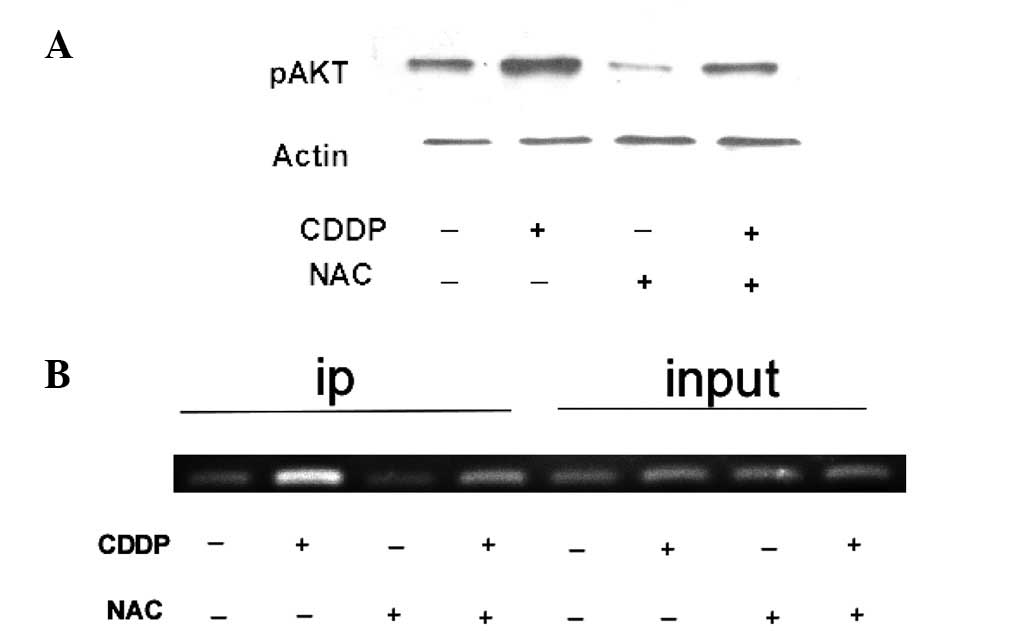
by STAT3, HCT-116 cells were treated with 60 mg/ml CDDP and 33
mg/ml NAC and the ChIP assay, which detects specific genomic DNA
sequences that are associated with particular transcription factors
in intact cells, was performed. HCT-116 cells were treated with 60
mg/ml of CDDP and 33 mg/ml NAC and immunoprecipitated with a STAT3
antibody. As shown in Fig. 4B, the
STAT3 bound chromatin was subjected to PCR using oligonucleotide
primers to amplify the region of the STAT3-binding site within the
AKT promoter. As shown in Fig. 4A,
NAC and to CDDP exposure were compared. In this experiment, CDDP
treatment reduced the AKT expression levels compared with cultures
exposed to NAC. These results indicate that STAT3 is able to bind
to the AKT promoter and CDDP increases the AKT activation.
Discussion
Reducing chemotherapy drug resistance is a huge
clinical challenge. It is well established that chemotherapy
induces drug resistance in tumor cells, resulting in treatment
failure. In addition, hyperactivation of AKT has been detected when
cancer cells acquire chemoresistance (4,7). The
present study showed that CDDP stimulates the JAK2/STAT3 pathway,
which participates in inducing AKT gene expression. Previous
studies (4,16) have shown that CDDP resistance is
associated with AKT overexpression. The activation of AKT promotes
the development of resistance to chemotherapy treatment (17–19).
In the present study, JAK2, activated by CDDP-induced ROS, was
associated with STAT3 phosphorylation and the transactivation of a
STAT-targeted AKT gene promoter. Reducing the expression of JAK2
using siRNA reduced the AKT expression. These data suggest that the
JAK2/STAT3 pathway is a potential target for overcoming
chemoresistance.
CDDP stimulates AKT activity in HCT-116 cells, with
the peak activity occurring with 200 μmol/l at 6 h.
Inhibition of ROS activity by NAC treatment partially inhibits
CDDP-stimulated JAK2 and STAT3 activities (Fig. 3A). Based on these findings, it is
proposed that ROS may have effects upstream of the JAK2/STAT3
pathway. Alterations to AKT at the protein level have been reported
in certain gastric cancers (20).
However, small number of tumors have exhibited elevated AKT mRNA
levels, which indicates that AKT is regulated at the
transcriptional level. Translational regulation of AKT has been
well documented in a number of studies (21–23).
In the present study, a sequence was selected from the human AKT
promoter and a STAT3 binding site was revealed within the promoter.
It was notable that STAT3 was not able to bind to the AKT promoter,
as revealed by the ChIP assay. The promoter of AKT activity was
significantly upregulated by CDDP. When ROS activity was inhibited,
the expression of AKT decreased in the HCT-116 cells. The AKT and
JAK2/STAT3 pathways are important in cellular processes associated
with chemoresistance (24,25). The results showed that the
JAK2/STAT3 pathway is able to mediate AKT expression and represents
a novel target for overcoming CDDP resistance in human tumors.
The present results indicate that the AKT promoter
construct containing the functional STAT3-binding site was
activated by CDDP-induced ROS, which are blocked by treatment with
NAC. JAK2-mediated phosphorylation of STAT3 is therefore required
for AKT promoter activation by ROS. Significant AKT activation was
observed in HCT-116 cells treated with CDDP. The observation that
AKT expression is enhanced by the JAK2/STAT3 pathway in HCT-116
cells treated with CDDP suggests that the JAK2/STAT3 pathway is a
potential target for overcoming chemoresistance.
In conclusion, the present study demonstrated that
STAT3 transcriptionally regulates the AKT gene. Blocking ROS with
NAC decreases AKT expression. These findings are important for two
reasons. Firstly, they provide a mechanistic understanding of the
upregulation of AKT. Secondly, the JAK2/STAT3 pathway was also
shown to mediate AKT expression, representing a potential target
for overcoming CDDP resistance in human tumors.
Acknowledgements
The authors would like to thank Mr.
Hong Xia for his outstanding administrative support and excellent
technical assistance in the present study. The study was supported
by grants from the National Natural Science Foundation of China
(No. 30770967) and Program for New Century Talent of the National
Ministry of Education, China.
References
|
1
|
Monzon FA, Ogino S, Hammond ME, Halling
KC, Bloom KJ and Nikiforova MN: The role of KRAS mutation testing
in the management of patients with metastatic colorectal cancer.
Arch Pathol Lab Med. 133:1600–1606. 2009.PubMed/NCBI
|
|
2
|
Brozovic A, Fritz G, Christmann M,
Zisowsky J, Jaehde U, Osmak M and Kaina B: Long-term activation of
SAPK/JNK, p38 kinase and Fas-L expression by cisplatin is
attenuated in human carcinoma cells that acquired drug resistance.
Int J Cancer. 112:974–985. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sánchez-Perez I, Murguía JR and Perona R:
Cisplatin induces a persistent activation of JNK that is related to
cell death. Oncogene. 16:533–540. 1998.PubMed/NCBI
|
|
4
|
Liu LZ, Zhou XD, Qian G, Shi X, Fang J and
Jiang BH: AKT1 amplification regulates cisplatin resistance in
human lung cancer cells through the mammalian target of
rapamycin/p70S6K1 pathway. Cancer Res. 67:6325–6332. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Toker A and Yoeli-Lerner M: Akt signaling
and cancer: surviving but not moving on. Cancer Res. 66:3963–3966.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bellacosa A, Kumar CC, Di Cristofano A and
Testa JR: Activation of AKT kinases in cancer: implications for
therapeutic targeting. Adv Cancer Res. 94:29–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deng R, Tang J, Xie BF, Feng GK, Huang YH,
Liu ZC and Zhu XF: SYUNZ-16, a newly synthesized alkannin
derivative, induces tumor cells apoptosis and suppresses tumor
growth through inhibition of PKB/AKT kinase activity and blockade
of AKT/FOXO signal pathway. Int J Cancer. 127:220–229. 2010.
View Article : Google Scholar
|
|
9
|
Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li
JH, Xu XM, Liu S, Chen J, Liu F, et al: Phosphoinositide
3-kinase/Akt pathway plays an important role in chemoresistance of
gastric cancer cells against etoposide and doxorubicin induced cell
death. Int J Cancer. 122:433–443. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang X, Fraser M, Moll UM, Basak A and
Tsang BK: Akt-mediated cisplatin resistance in ovarian cancer:
modulation of p53 action on caspase-dependent mitochondrial death
pathway. Cancer Res. 66:3126–3136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fraser M, Leung BM, Yan X, Dan HC, Cheng
JQ and Tsang BK: p53 is a determinant of X-linked inhibitor of
apoptosis protein/Akt-mediated chemoresistance in human ovarian
cancer cells. Cancer Res. 63:7081–7088. 2003.PubMed/NCBI
|
|
12
|
Pellegrini S and Dusanter-Fourt I: The
structure, regulation and function of the Janus kinases (JAKs) and
the signal transducers and activators of transcription (STATs). Eur
J Biochem. 248:615–633. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun J, Blaskovich MA, Jove R, Livingston
SK, Coppola D and Sebti SM: Cucurbitacin Q: a selective STAT3
activation inhibitor with potent antitumor activity. Oncogene.
24:3236–3245. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park S, Kim D, Kaneko S, Szewczyk KM,
Nicosia SV, Yu H, Jove R and Cheng JQ: Molecular cloning and
characterization of the human AKT1 promoter uncovers its
up-regulation by the Src/Stat3 pathway. J Biol Chem.
280:38932–38941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee BL, Lee HS, Jung J, Cho SJ, Chung HY,
Kim WH, Jin YW, Kim CS and Nam SY: Nuclear factor-kappaB activation
correlates with better prognosis and Akt activation in human
gastric cancer. Clin Cancer Res. 11:2518–2525. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Testa JR and Bellacosa A: AKT plays a
central role in tumorigenesis. Proc Natl Acad Sci U S A.
98:10983–10985. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ruggero D and Sonenberg N: The Akt of
translational control. Oncogene. 24:7426–7434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim D, Dan HC, Park S, Yang L, Liu Q,
Kaneko S, Ning J, He L, Yang H, Sun M, et al: AKT/PKB signaling
mechanisms in cancer and chemoresistance. Front Biosci. 10:975–987.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang HL, Fang LW, Lu SP, Chou CK, Luh TY
and Lai MZ: DNA-damaging reagents induce apoptosis through reactive
oxygen species-dependent Fas aggregation. Oncogene. 22:8168–8177.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Antico Arciuch VG, Galli S, Franco MC, Lam
PY, Cadenas E, Carreras MC and Poderoso JJ: Akt1 intramitochondrial
cycling is a crucial step in the redox modulation of cell cycle
progression. PLoS One. 4:e75232009.PubMed/NCBI
|
|
22
|
Carpten JD, Faber AL, Horn C, Donoho GP,
Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage
S, et al: A transforming mutation in the pleckstrin homology domain
of AKT1 in cancer. Nature. 448:439–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu X, Shi Y, Han EK, Chen Z, Rosenberg
SH, Giranda VL, Luo Y and Ng SC: Down regulation of Akt1 inhibits
anchorage-independent cell growth and induces apoptosis in cancer
cells. Neoplasia. 3:278–286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Real PJ, Sierra A, De Juan A, Segovia JC,
Lopez-Vega JM and Fernandez-Luna JL: Resistance to chemotherapy via
Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer
cells. Oncogene. 21:7611–7618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Garcia R, Bowman TL, Niu G, Yu H, Minton
S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, et
al: Constitutive activation of Stat3 by the Src and JAK tyrosine
kinases participates in growth regulation of human breast carcinoma
cells. Oncogene. 20:2499–2513. 2001. View Article : Google Scholar : PubMed/NCBI
|