Introduction
Colorectal cancer (CRC) is one of the most common
types of cancer in the world. Despite advances in chemotherapeutic
agents, the prognosis for patients with metastatic CRC (mCRC)
remains poor (1). Cetuximab (Cmab)
is a chimeric monoclonal antibody (moAb) for epidermal growth
factor receptor (EGFR), and has been shown to be effective for mCRC
in combination with chemotherapy or as a single agent (2–6). EGFR
is expressed in various malignancies, including CRC (7). EGFR activation plays an important role
in growth and progression, involving proliferation, angiogenesis,
invasion and metastasis (8). Cmab
binds to the extracellular domain of EGFR and inhibits downstream
signal transduction (9). However,
only 10–20% of patients with mCRC respond to Cmab (3). The identification of biomarkers of
response to Cmab for mCRC is important in the selection of mCRC
patients who should be administered Cmab to avoid unnecessary
toxicities and ineffective, expensive therapy. Analysis of clinical
trials for mCRC indicates that KRAS mutation is a negative
predictor of Cmab-based therapies (10–14).
KRAS belongs to the oncogene family of genes and is
activated by EGFR which binds to a ligand (8). KRAS mutation continuously
activates downstream RAS-RAF-MAPK pathways whether EGFR is
activated or blocked by the antibody (8). Although KRAS mutation may be
considered a highly specific negative biomarker of response, it is
also poorly sensitive (15). The
identification of additional biomarkers is necessary to improve
sensitivity. EGFR copy number (16–18),
the levels of expression of amphiregulin and epiregulin (19), FCGR2A and FCGR3A polymorphisms
(20), BRAF mutation, PIK3CA
mutation and PTEN inactivation (18,21–26)
have been reported to be associated with response to Cmab, but at
present, these markers cannot be used to select patients who are
eligible for Cmab treatment.
A recent study revealed that p53 mutations
are predictive of Cmab sensitivity (27). Another study reported that Ki67
expression is downregulated following Cmab-based neoadjuvant
chemoradiotherapy in rectal cancer (28). Moreover, it has been reported that
expression of E-cadherin is a marker of response to Cmab in
vitro(29). In the present
study, we examined the expression of p53, Ki67 and E-cadherin
together with KRAS status and assessed their predictive
value as biomarkers of response to Cmab in mCRC.
Materials and methods
Patients and tissue samples
We assessed 36 mCRC patients treated with Cmab-based
therapy, who had tumor tissues available for molecular analysis.
Tumor response was evaluated according to the Response Evaluation
Criteria in Solid Tumors (RECIST). Patient tumor response was
classified as complete response (CR), partial response (PR), stable
disease (SD) or progressive disease (PD). Patients who achieved PR
or CR or SD were considered responders (controlled disease; CD).
Patients who achieved PD were considered non-responders.
Follow-up was performed on a clinical basis and CT scan until
disease progression, mortality or the last follow-up point at which
data were monitored. The study was conducted in accordance with the
Helsinki Declaration and was approved by the Ethics Committee of
Osaka City University, Osaka, Japan. Informed consent was obtained
from all patients or guardians.
DNA extraction
DNA was extracted from tissue sections fixed in 10%
buffered formalin and embedded in paraffin. An adjacent section
stained with hematoxylin and eosin was used as a guide in the
selection of areas for microdissection under a dissecting
microscope, using a sterile scalpel blade. Genomic DNA was
extracted from the paraffin-embedded tissue using Proteinase K
(Gibco-BRL, Gaithersburg, MD, USA).
Dot-blot hybridization
The DNA was amplified using a heminested PCR
protocol as previously described (30). PCR amplification of exon 2 of a
KRAS-containing codons 12 and 13 was first performed using
the following primers: forward,
5’-CGTCCACAAAATGATTCTGAATTAGCTGTATC-3’ and reverse,
5’-CCTTATGTGTGACATGTTCTAATATAGT CAC-3’. Thirty-five cycles (92°C
for 30 sec and 67°C for 30 sec) were performed, followed by a
10-min extension at 72°C. Initial PCR products were diluted and
further amplified using a new forward primer,
5’-AGGCCTGCTGAAAATGAC-3’, and the same reverse primer described
above. Thirty-five cycles (92°C for 25 sec, 55°C for 25 sec and
72°C for 25 sec) were performed, followed by a 10-min extension at
72°C. The 104-bp amplicons were then dot-blotted onto nylon filters
(Hybond-N; Amersham, Buckinghamshire, UK) and hybridized with
radiolabeled oligomer primers representing all possible mutations
at codon 12 and the GAC mutation of codon 13. Direct sequencing was
performed to confirm the presence of KRAS mutations at codons 12
and 13, which were detected by dot-blot hybridization.
Immunohistochemical study
All tissues were fixed in 10% formalin immediately
after surgical resection or biopsy and embedded in paraffin. The
slides were deparaffinized and heated for 10 min at 105°C by
autoclave in Target Retrieval Solution (Dako, Carpinteria, CA,
USA). Sections were then incubated with 3% hydrogen peroxide to
block endogenous peroxidase activity. Thereafter, sections were
incubated in 10% normal goat or rabbit serum to reduce non-specific
antibody binding. Primary monoclonal antibodies were directed
against p53 (DO7, dilution 1:50; Dako), Ki67 (MIB-1, dilution 1:50;
Dako) and E-cadherin (clone NCH-38, dilution 1:200; Dako). Tissue
sections were incubated with each antibody overnight at 4°C. After
washing in phosphate-buffered saline (PBS), tissues were incubated
with horseradish peroxidase-conjugated anti-rabbit or anti-mouse Ig
polymer as a secondary antibody (Envision kit; Dako) for 30 min at
room temperature, according to the manufacturer’s instructions. The
slides were treated with streptavidin-peroxidase reagent and
incubated in PBS and diaminobenzidine and 1% hydrogen peroxide v/v,
followed by counterstaining with Mayer’s hematoxylin. Positive and
negative controls for each marker were used according to the
manufacturer’s instructions (Dako). The immunostained slides were
independently examined and scored by two investigators.
Immunohistochemical scoring was performed in a blind manner. p53
expression was semi-quantitatively analyzed according to the
percentage of cells showing nuclear positivity: 0, 0 to 10%; 1+,
>10 to 25%; 2+, >25% to 50%; 3+, >50%. According to
previous studies, p53 expression was considered positive when
scores were >1, and negative when scores were 0 (31–34).
For the tissue evaluation of Ki67, each slide was scored based on
the percentage of positively stained malignant nuclei. According to
the recommended classification in previous studies, the cut-off
Ki67 positivity was >40% positive tumor cells with nuclear
staining (32,33,35).
E-cadherin antibody stained the membrane intensely and the
cytoplasm of cancer cells weakly. E-cadherin expression was
semi-quantitatively analyzed according to the percentage of cells
showing membrane positivity: 0, 0%; 1+, >0 to 25%; 2+, >25 to
50%; 3+, >50%. According to previous studies, E-cadherin
expression was considered positive when scores were >1 and
negative when scores were 0 (36,37). A
case with cytoplasmic staining only was determined as
E-cadherin-negative.
Statistical analysis
Statistical analysis was performed using SPSS 13.0
statistical software (SPSS Inc., Chicago, IL, USA). We examined the
association between the biological parameter status and treatment
response using Chi-square analysis. We also estimated odds ratios
(ORs) using logistic regression analysis. All P-values were 2-sided
and P<0.05 was considered to indicate a statistically
significant difference. Cut-off values for different biomarkers
included in this study were selected before statistical
analysis.
Results
A total of 36 mCRC patients treated with Cmab-based
therapy, including 24 males and 12 females with a mean age of 62.2
years (range, 29–79) were included in this study (Table I). Twenty-seven patients received
Cmab-based therapy as third line therapy, 8 patients received it as
second line and only one patient received it as first line therapy.
With regard to concurrent chemotherapy, 17 patients received Cmab
with irinotecan, 18 received Cmab alone and one patient received
Cmab with mFOLFOX6. Response to Cmab therapy demonstrated that 19
(53%) patients had a CD (8 PR and 11 SD), while 17 (47%) were in
PD.
 | Table IPatient characteristics. |
Table I
Patient characteristics.
|
Characteristics | No. | % |
|---|
| No. of
patients | 36 | |
| Age (years) | | |
| Median | 62.2 | |
| Range | 29–79 | |
| Gender | | |
| Male | 24 | 67 |
| Female | 12 | 33 |
| Site of tumor | | |
| Colon | 20 | 56 |
| Rectum | 16 | 44 |
| Synchronous
metastasis | 23 | 66 |
| Metachronous
recurrence | 22 | 63 |
| Lines of
treatment | | |
| ≤2 | 9 | 25 |
| 3 | 27 | 75 |
| Concurrent
chemotherapy | | |
| Yes | 18 | 50 |
| No | 18 | 50 |
KRAS status analysis
Table II shows the
results of KRAS status analysis. Results of dot-blot
hybridization were equivalent to that of direct sequencing.
KRAS was mutated in 12 (33%) of 36 tumors. Ten (83%) of 12
tumors had KRAS mutation in codon 12, while 2 (17%) of 12
had mutations in codon 13.
| Table IIKRAS mutation types. |
Table II
KRAS mutation types.
| Types of mutations
found in codon 12a | Codon 13a | Total |
|---|
|
|
|---|
| Asp (GAT) | Val (GTT) | Ser (AGT) | Arg (CGT) | Cys (TGT) | Ala (GCT) | Asp (GTC) |
|---|
| Number of tumors
with each KRAS mutation/number of tumors with KRAS mutation
(%) | 5/12 (42) | 4/12 (33) | 1/12 (8) | 0/12 (0) | 0/12 (0) | 0/12 (0) | 2/12 (17) | 12/36a |
Expression of p53, Ki67 and E-cadherin
demonstrated by immunohistochemistry (IHC)
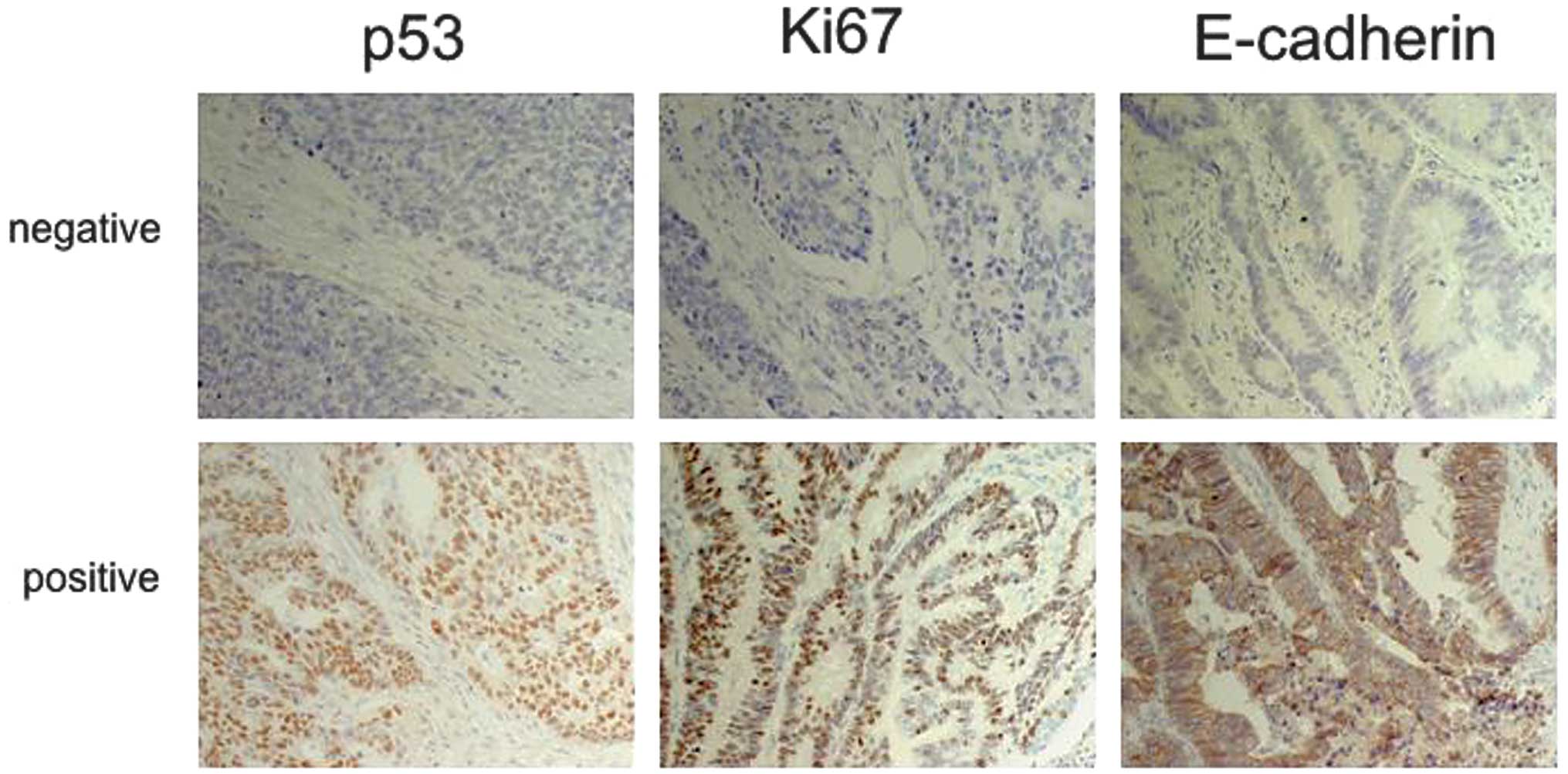
Fig. 1 shows the
expression of p53, Ki67 and E-cadherin. p53, Ki67 and E-cadherin
were positive in 29 (81%), 21 (58%), and 22 (61%) of 36 tumors,
respectively.
Correlation between response to Cmab and
KRAS status, and expression of p53, Ki67 and E-cadherin
Table III shows the
response to treatment with Cmab according to KRAS status,
and expression of p53, Ki67 and E-cadherin. Sixteen (67%) of 24
patients with KRAS wild-type tumors were found in responders
compared with 3 (25%) of 12 patients with KRAS mutant-type
tumors in responders. Seventeen (77%) of 22 patients with
E-cadherin-positive tumors were found in responders, compared with
2 (14%) of 14 patients with E-cadherin-negative tumors found in
responders. KRAS status and expression of E-cadherin were
significantly associated with response to Cmab treatment (P=0.033
and P<0.001, respectively). Expression of p53 and Ki67 were not
associated with response to Cmab treatment (P=0.219 and P=1.000,
respectively).
| Table IIIResponse to treatment according to
KRAS status and p53, Ki67 and E-cadherin IHC. |
Table III
Response to treatment according to
KRAS status and p53, Ki67 and E-cadherin IHC.
| Responder | Non-responder | P-value |
|---|
| KRAS status | | | |
| Wild-type | 16 | 8 | 0.033a |
| Mutant | 3 | 9 | |
| p53 IHC | | | |
| Positive | 17 | 12 | 0.219 |
| Negative | 2 | 5 | |
| Ki67 IHC | | | |
| Positive | 11 | 10 | 1.000 |
| Negative | 8 | 7 | |
| E-cadherin IHC | | | |
| Positive | 17 | 2 | <0.001a |
| Negative | 5 | 12 | |
Expression of E-cadherin in KRAS
wild-type patients
E-cadherin was positive in 14 (58%) of 24
KRAS wild-type tumors. Fourteen (93%) of 15 patients with
E-cadherin-positive tumors were found in responders compared with 2
(22%) of 9 patients with E-cadherin-negative tumors found in
responders. Expression of E-cadherin was significantly associated
with response to Cmab treatment in KRAS wild-type patients
(P=0.001; Table IV).
| Table IVResponse to treatment according to
combined KRAS status and E-cadherin IHC. |
Table IV
Response to treatment according to
combined KRAS status and E-cadherin IHC.
| E-cadherin IHC | Responder | Non-responder | P-value |
|---|
| KRAS wild-type | | | |
| Positive | 14 | 1 | 0.001a |
| Negative | 2 | 7 | |
| KRAS mutant
type | | | |
| Positive | 3 | 4 | 0.205 |
| Negative | 0 | 5 | |
Expression of E-cadherin in KRAS
mutant-type patients
E-cadherin was positive in 7 (58%) of 12 KRAS
mutant-type tumors. Expression of E-cadherin was not significantly
associated with response to Cmab treatment in KRAS
mutant-type patients (P=0.205). However, all 3 responders with
KRAS mutant-type tumors expressed E-cadherin (Table IV).
Univariate and multivariate models
In the univariate analysis, which included age
(<62 vs. ≥62 years), gender (male vs. female), site of tumors
(colon vs. rectum), concurrent chemotherapy (yes vs. no),
KRAS status (wild-type vs. mutant), expression of p53, Ki67
and E-cadherin with IHC (positive vs. negative), only KRAS
status and expression of E-cadherin demonstrated a significant
association with response to treatment with Cmab. In the
multivariate analysis, KRAS status and E-cadherin IHC
significantly affected the efficacy of Cmab-based therapy
(KRAS: OR, 20.83; 95% CI, 1.80–241.18; P=0.015; E-cadherin:
OR, 54.91; 95%CI, 4.53–664.89; P=0.002; Table V). No evidence of interaction
between KRAS status and expression of E-cadherin was
detected.
| Table VUnivariate and multivariate analysis
with respect to the efficacy of Cmab. |
Table V
Univariate and multivariate analysis
with respect to the efficacy of Cmab.
| Variable | Univariate | Multivariate |
|---|
|
|
|---|
| P-value | Odds ratio | 95% CI | P-value |
|---|
| KRAS status | | | | |
| Wild-type vs.
mutant | 0.033 | 20.83 | 1.80–241.18 | 0.015a |
| E-cadherin | | | | |
| Positive vs.
negative | <0.001 | 54.91 | 4.53–664.89 | 0.002a |
Discussion
E-cadherin is a calcium-regulated homophilic
cell-cell adhesion molecule. Previous studies have reported that
E-cadherin regulates not only cell-cell adhesion, but also
intracellular signaling cascades, including the Akt and MAPK
pathways (38,39). It has been revealed that E-cadherin
coexists with EGFR in a complex and the extracellular domain of
E-cadherin regulates the ability of EGFR to respond to its ligand
(40). Furthermore, it has been
reported that there is a significant correlation between expression
of E-cadherin and sensitivity to the EGFR tyrosine kinase
inhibitor, gefitinib, in non-small cell lung cancer cell lines
(41), and that the most
gefitinib-sensitive cell lines have higher levels of EGFR
activation (42). In addition, loss
of E-cadherin has been demonstrated to be a marker of poor response
to the antiproliferative effect of Cmab in a panel of urothelial
carcinoma cell lines (29). These
findings suggest that cells expressing E-cadherin increase
dependence on EGFR for cell growth and survival and that cells
lacking E-cadherin have developed other activating mechanisms that
bypass EGFR signaling for cell growth and survival, and then
acquire resistance to EGFR inhibition. Based on these findings, we
hypothesized that the expression of E-cadherin, detected with IHC,
may be a biomarker of response to Cmab in mCRC. This is the first
study to investigate the correlation between the expression of
E-cadherin demonstrated by IHC and the effect of Cmab in mCRC
clinical specimen. In our experience, expression of E-cadherin
correlated with a higher controlled disease rate in mCRC treated
with Cmab.
Our results are consistent with the knowledge that
KRAS mutant-type is negative predictor of Cmab-based therapy in
mCRC. Previous studies have reported that the controlled disease
rate of mCRC-treated Cmab-based therapy were 48 to 83% (mean, 67%)
in patients with KRAS wild-type tumors and 10 to 74% (mean, 40%) in
patients with KRAS mutant-type tumors (14,19,21,43,44).
Our data also demonstrated that KRAS mutant-type tumors were
correlated with a lower controlled disease rate.
The p53 tumor suppressor gene has been demonstrated
to regulate cell cycle progression and apoptosis. p53 mutations are
found in 40 to 60% of patients with colorectal cancer (33). Mutated p53 protein accumulates in
the nucleus and is detected by IHC (33,45).
This method has since been suggested to predict p53 mutations. A
previous study has suggested that p53 mutations are predictive of
Cmab sensitivity, particularly in patients without KRAS mutation
(27). Therefore, we examined the
correlation between p53 expression using IHC and the efficacy of
Cmab-based therapy; however, no correlation was identified.
The Ki67 antigen recognizes the nuclei of
proliferating cells throughout the cell cycle, except during the G0
and early G1 phases (33). The Ki67
labeling index is associated with tumor proliferation (46). Recent studies have reported that
neoadjuvant chemoradiotherapy with Cmab decreased the levels of the
Ki67 labeling index in rectal cancer (28). We hypothesized that the Ki67
labeling index reflects the efficacy of Cmab-based therapy in CRC;
thus, we examined the correlation between Ki67 and the efficacy of
Cmab-based therapy; however, no correlation was found.
According to univariate and multivariate analysis,
the efficacy of Cmab was significantly associated with KRAS status
and E-cadherin expression. Moreover, multivariate analysis also
demonstrated that the two factors were independent predictors of
the efficacy of Cmab-based therapy in mCRC.
In the KRAS wild-type tumors, E-cadherin-positive
status was correlated with a higher controlled disease rate. When
expression of E-cadherin was considered a positive predictor of
Cmab in KRAS wild-type mCRC, both sensitivity and specificity were
87.5%. Moreover, in KRAS mutant-type tumors, all responders
expressed E-cadherin. Our results suggest that the expression of
E-cadherin may be a predictive marker of Cmab-based therapy in mCRC
independently or in combination with KRAS status analysis. Since
E-cadherin IHC is a comparatively simple method, it is easy to
introduce as a biomarker of Cmab-based therapy. In addition, it is
possible that the expression of E-cadherin may be predictive of
sensitivity to panitumumab, a fully human monoclonal antibody
targeting the EGFR. However, as our study had a small sample size
and was retrospective, it is necessary to conduct a large,
prospective clinical trial in order to confirm this finding. In
conclusion, our results indicate that detection of the expression
of E-cadherin via IHC may be a positive predictor of Cmab-based
therapy in mCRC, and that the combination of E-cadherin IHC and
KRAS analysis may be a more sensitive biomarker than KRAS analysis
alone.
References
|
1
|
Poston GJ, Figueras J, Giuliante F, et al:
Urgent need for a new staging system in advanced colorectal cancer.
J Clin Oncol. 26:4828–4833. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saltz LB, Meropol NJ, Loehrer PJ Sr,
Needle MN, Kopit J and Mayer RJ: Phase II trial of cetuximab in
patients with refractory colorectal cancer that expresses the
epidermal growth factor receptor. J Clin Oncol. 22:1201–1208. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cunningham D, Humblet Y, Siena S, et al:
Cetuximab mono-therapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
51:337–345. 2004. View Article : Google Scholar
|
|
4
|
Chung KY, Shia J, Kemeny NE, et al:
Cetuximab shows activity in colorectal cancer patients with tumors
that do not express the epidermal growth factor receptor by
immunohistochemistry. J Clin Oncol. 23:1803–1810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jonker DJ, O’Callaghan CJ, Karapetis CS,
et al: Cetuximab for the treatment of colorectal cancer. N Engl J
Med. 357:2040–2048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sobrero AF, Maurel J, Fehrenbacher L, et
al: EPIC: phase III trial of cetuximab plus irinotecan after
fluoropyrimidine and oxaliplatin failure in patients with
metastatic colorectal cancer. J Clin Oncol. 26:2311–2319. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Camp ER, Summy J, Bauer TW, Liu W, Gallick
GE and Ellis LM: Molecular mechanisms of resistance to therapies
targeting the epidermal growth factor receptor. Clin Cancer Res.
11:397–405. 2005.PubMed/NCBI
|
|
8
|
Siena S, Sartore-Bianchi A, Di
Nicolantonio F, Balfour J and Bardelli A: Biomarkers predicting
clinical outcome of epidermal growth factor receptor-targeted
therapy in metastatic colorectal cancer. J Natl Cancer Inst.
101:1308–1324. 2009. View Article : Google Scholar
|
|
9
|
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ,
Kussie P and Ferguson KM: Structural basis for inhibition of the
epidermal growth factor receptor by cetuximab. Cancer Cell.
7:301–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Van Cutsem E, Kohne CH, Hitre E, et al:
Cetuximab and chemo-therapy as initial treatment for metastatic
colorectal cancer. N Engl J Med. 360:1408–1417. 2009.PubMed/NCBI
|
|
11
|
Bokemeyer C, Bondarenko I, Makhson A, et
al: Fluorouracil, leucovorin, and oxaliplatin with and without
cetuximab in the first-line treatment of metastatic colorectal
cancer. J Clin Oncol. 27:663–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
et al: K-ras mutations and benefit from cetuximab in advanced
colorectal cancer. N Engl J Med. 359:1757–1765. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lièvre A, Bachet JB, Boige V, et al: KRAS
mutations as an independent prognostic factor in patients with
advanced colorectal cancer treated with cetuximab. J Clin Oncol.
26:374–379. 2008.
|
|
14
|
Di Fiore F, Blanchard F, Charbonnier F, et
al: Clinical relevance of KRAS mutation detection in metastatic
colorectal cancer treated by Cetuximab plus chemotherapy. Br J
Cancer. 96:1166–1169. 2007.PubMed/NCBI
|
|
15
|
Bardelli A and Siena S: Molecular
mechanisms of resistance to cetuximab and panitumumab in colorectal
cancer. J Clin Oncol. 28:1254–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cappuzzo F, Finocchiaro G, Rossi E, et al:
EGFR FISH assay predicts for response to cetuximab in chemotherapy
refractory colorectal cancer patients. Ann Oncol. 19:717–723. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Personeni N, Fieuws S, Piessevaux H, et
al: Clinical usefulness of EGFR gene copy number as a predictive
marker in colorectal cancer patients treated with cetuximab: a
fluorescent in situ hybridization study. Clin Cancer Res.
14:5869–5876. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frattini M, Saletti P, Romagnani E, et al:
PTEN loss of expression predicts cetuximab efficacy in metastatic
colorectal cancer patients. Br J Cancer. 97:1139–1145. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Khambata-Ford S, Garrett CR, Meropol NJ,
et al: Expression of epiregulin and amphiregulin and K-ras mutation
status predict disease control in metastatic colorectal cancer
patients treated with cetuximab. J Clin Oncol. 25:3230–3237. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang W, Gordon M, Schultheis AM, et al:
FCGR2A and FCGR3A polymorphisms associated with clinical outcome of
epidermal growth factor receptor expressing metastatic colorectal
cancer patients treated with single-agent cetuximab. J Clin Oncol.
25:3712–3718. 2007. View Article : Google Scholar
|
|
21
|
Benvenuti S, Sartore-Bianchi A, Di
Nicolantonio F, et al: Oncogenic activation of the RAS/RAF
signaling pathway impairs the response of metastatic colorectal
cancers to anti-epidermal growth factor receptor antibody
therapies. Cancer Res. 67:2643–2648. 2007. View Article : Google Scholar
|
|
22
|
Di Nicolantonio F, Martini M, Molinari F,
et al: Wild-type BRAF is required for response to panitumumab or
cetuximab in metastatic colorectal cancer. J Clin Oncol.
26:5705–5712. 2008.
|
|
23
|
Sartore-Bianchi A, Martini M, Molinari F,
et al: PIK3CA mutations in colorectal cancer are associated with
clinical resistance to EGFR-targeted monoclonal antibodies. Cancer
Res. 69:1851–1857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jhawer M, Goel S, Wilson AJ, et al: PIK3CA
mutation/PTEN expression status predicts response of colon cancer
cells to the epidermal growth factor receptor inhibitor cetuximab.
Cancer Res. 68:1953–1961. 2008. View Article : Google Scholar
|
|
25
|
Perrone F, Lampis A, Orsenigo M, et al:
PI3KCA/PTEN deregulation contributes to impaired responses to
cetuximab in metastatic colorectal cancer patients. Ann Oncol.
20:84–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Loupakis F, Pollina L, Stasi I, et al:
PTEN expression and KRAS mutations on primary tumors and metastases
in the prediction of benefit from cetuximab plus irinotecan for
patients with meta-static colorectal cancer. J Clin Oncol.
27:2622–2629. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oden-Gangloff A, Di Fiore F, Bibeau F, et
al: TP53 mutations predict disease control in metastatic colorectal
cancer treated with cetuximab-based chemotherapy. Br J Cancer.
100:1330–1335. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Debucquoy A, Haustermans K, Daemen A, et
al: Molecular response to cetuximab and efficacy of preoperative
cetuximab-based chemoradiation in rectal cancer. J Clin Oncol.
27:2751–2757. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Black PC, Brown GA, Inamoto T, et al:
Sensitivity to epidermal growth factor receptor inhibitor requires
E-cadherin expression in urothelial carcinoma cells. Clin Cancer
Res. 14:1478–1486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yashiro M, Carethers JM, Laghi L, et al:
Genetic pathways in the evolution of morphologically distinct
colorectal neoplasms. Cancer Res. 61:2676–2683. 2001.PubMed/NCBI
|
|
31
|
Lenz HJ, Danenberg KD, Leichman CG, et al:
p53 and thymidylate synthase expression in untreated stage II colon
cancer: associations with recurrence, survival, and site. Clin
Cancer Res. 4:1227–1234. 1998.PubMed/NCBI
|
|
32
|
Allegra CJ, Paik S, Colangelo LH, et al:
Prognostic value of thymidylate synthase, Ki-67, and p53 in
patients with Dukes’ B and C colon cancer: a National Cancer
Institute-National Surgical Adjuvant Breast and Bowel Project
collaborative study. J Clin Oncol. 21:241–250. 2003.
|
|
33
|
Jakob C, Liersch T, Meyer W, Becker H,
Baretton GB and Aust DE: Predictive value of Ki67 and p53 in
locally advanced rectal cancer: correlation with thymidylate
synthase and histo-pathological tumor regression after neoadjuvant
5-FU-based chemoradiotherapy. World J Gastroenterol. 14:1060–1066.
2008. View Article : Google Scholar
|
|
34
|
El-Serafi MM, Bahnassy AA, Ali NM, et al:
The prognostic value of c-Kit, K-ras codon 12, and p53 codon 72
mutations in Egyptian patients with stage II colorectal cancer.
Cancer. 116:4954–4964. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Triest B, Pinedo HM, Blaauwgeers JL,
et al: Prognostic role of thymidylate synthase, thymidine
phosphorylase/platelet-derived endothelial cell growth factor, and
proliferation markers in colorectal cancer. Clin Cancer Res.
6:1063–1072. 2000.
|
|
36
|
Dorudi S, Sheffield JP, Poulsom R,
Northover JM and Hart IR: E-cadherin expression in colorectal
cancer. An immunocytochemical and in situ hybridization study Am J
Pathol. 142:981–986. 1993.PubMed/NCBI
|
|
37
|
Lugli A, Zlobec I, Minoo P, et al:
Prognostic significance of the wnt signalling pathway molecules
APC, beta-catenin and E-cadherin in colorectal cancer: a tissue
microarray-based analysis. Histopathology. 50:453–464. 2007.
View Article : Google Scholar
|
|
38
|
Pece S, Chiariello M, Murga C and Gutkind
JS: Activation of the protein kinase Akt/PKB by the formation of
E-cadherin-mediated cell-cell junctions. Evidence for the
association of phosphatidylinositol 3-kinase with the E-cadherin
adhesion complex. J Biol Chem. 274:19347–19351. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pece S and Gutkind JS: Signaling from
E-cadherins to the MAPK pathway by the recruitment and activation
of epidermal growth factor receptors upon cell-cell contact
formation. J Biol Chem. 275:41227–41233. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fedor-Chaiken M, Hein PW, Stewart JC,
Brackenbury R and Kinch MS: E-cadherin binding modulates EGF
receptor activation. Cell Commun Adhes. 10:105–118. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Witta SE, Gemmill RM, Hirsch FR, et al:
Restoring E-cadherin expression increases sensitivity to epidermal
growth factor receptor inhibitors in lung cancer cell lines. Cancer
Res. 66:944–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lièvre A, Bachet JB, Le Corre D, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006.PubMed/NCBI
|
|
44
|
De Roock W, Piessevaux H, De Schutter J,
et al: KRAS wild-type state predicts survival and is associated to
early radiological response in metastatic colorectal cancer treated
with cetuximab. Ann Oncol. 19:508–515. 2008.PubMed/NCBI
|
|
45
|
Noske A, Lipka S, Budczies J, et al:
Combination of p53 expression and p21 loss has an independent
prognostic impact on sporadic colorectal cancer. Oncol Rep. 22:3–9.
2009.PubMed/NCBI
|
|
46
|
Saleh HA, Jackson H, Khatib G and Banerjee
M: Correlation of bcl-2 oncoprotein immunohistochemical expression
with proliferation index and histopathologic parameters in
colorectal neoplasia. Pathol Oncol Res. 5:273–279. 1999. View Article : Google Scholar
|