Introduction
Malignant rhabdoid tumors of the kidney (MRTKs) are
uncommon renal tumors which mainly occur in children and are
extremely rare in adult patients. The present study describes a
malignant rhabdoid tumor identified in a patient’s left kidney and
the diagnosis which was made mainly based on characteristic
histological findings and immunohistochemical features. To the
authors’ knowledge, only five adult cases have been reported in the
English-language literature and the present case is the first
report of adult MRTK in China (1–5). We
propose that this study is likely play a significant role in
guiding clinical practice in the diagnosis and therapy of
MRTKs.
The study was approved by the ethics committee of
the Second Hospital of Tianjin Medical University, Tianjin
Institute of Urology (Tianjin, China). Informed consent was
obtained from the patient prior to the study.
Case report
A 59-year-old male patient was admitted to the
Second Hospital of Tianjin Medical University due to an
asymptomatic mass located in the left kidney which was revealed by
ultrasonography one week before. The patient had no other symptoms
with the exception of a weight loss of 6 kg over the past months.
Physical and laboratory examination revealed no abnormalities.
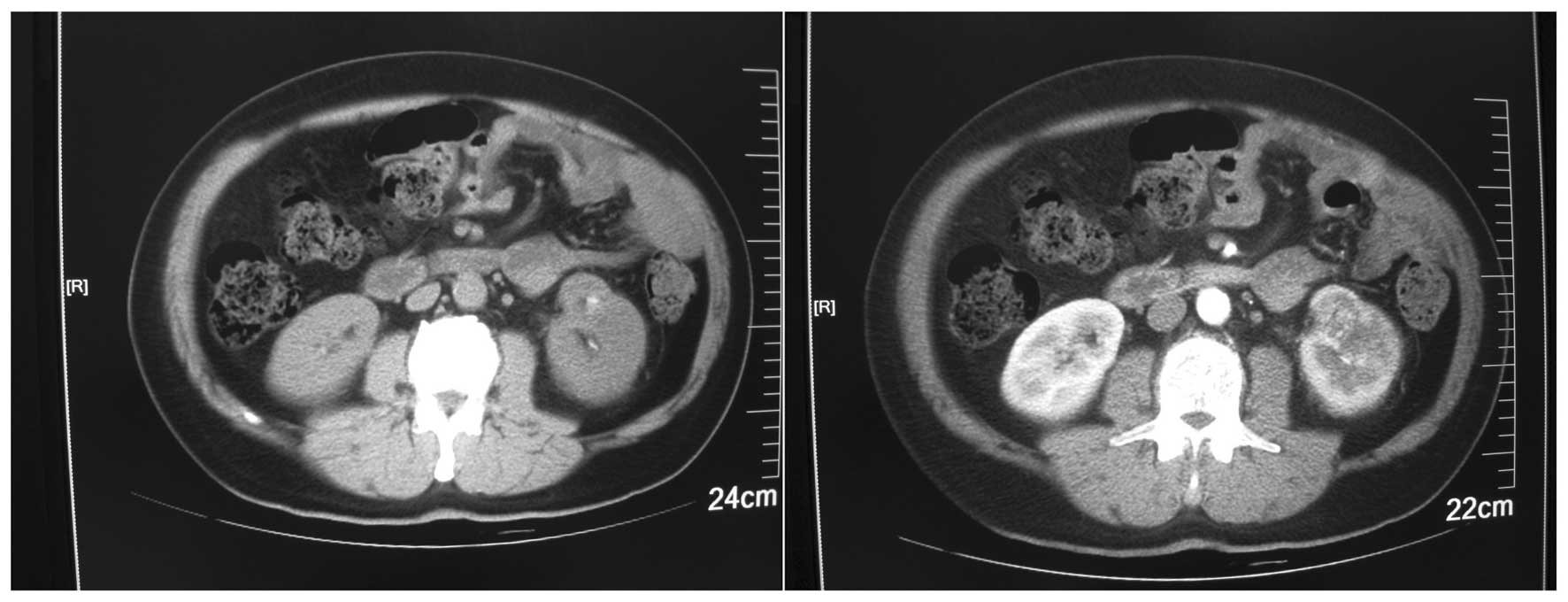
Ultrasonography revealed the presence of a mass, measuring
4.9×3.8×5.0 cm in the left kidney, with medium echo, as well as an
unclear boundary and internal inhomogeneous echo. A computed
tomography scan revealed a mixed density renal mass in the lower
pole of the left kidney, measuring 5×4×5 cm. The lesion exhibited
clear peripheral enhancement at the cortical phase and a low
density at the parenchymal and delayed phase. There was also
increased density in the left perirenal fat. In addition, the
following were observed: a shadow with liquid density in the left
pleural cavity, nodules with ring-enhancement posterior to the left
costophrenic angle and lymph node enlargement around the left
abdominal aorta (Fig. 1). A chest
radiograph revealed a group of flakes with increased density in the
left lung field.
An initial diagnosis of a space-occupying lesion of
the left kidney, retroperitoneal and left costophrenic angle lymph
node metastases and a space-occupying lesion of the left lung was
proposed (with suspicions of renal cell carcinoma; the clinical TNM
staging was T3aN2M1) and subsequently, the patient underwent left
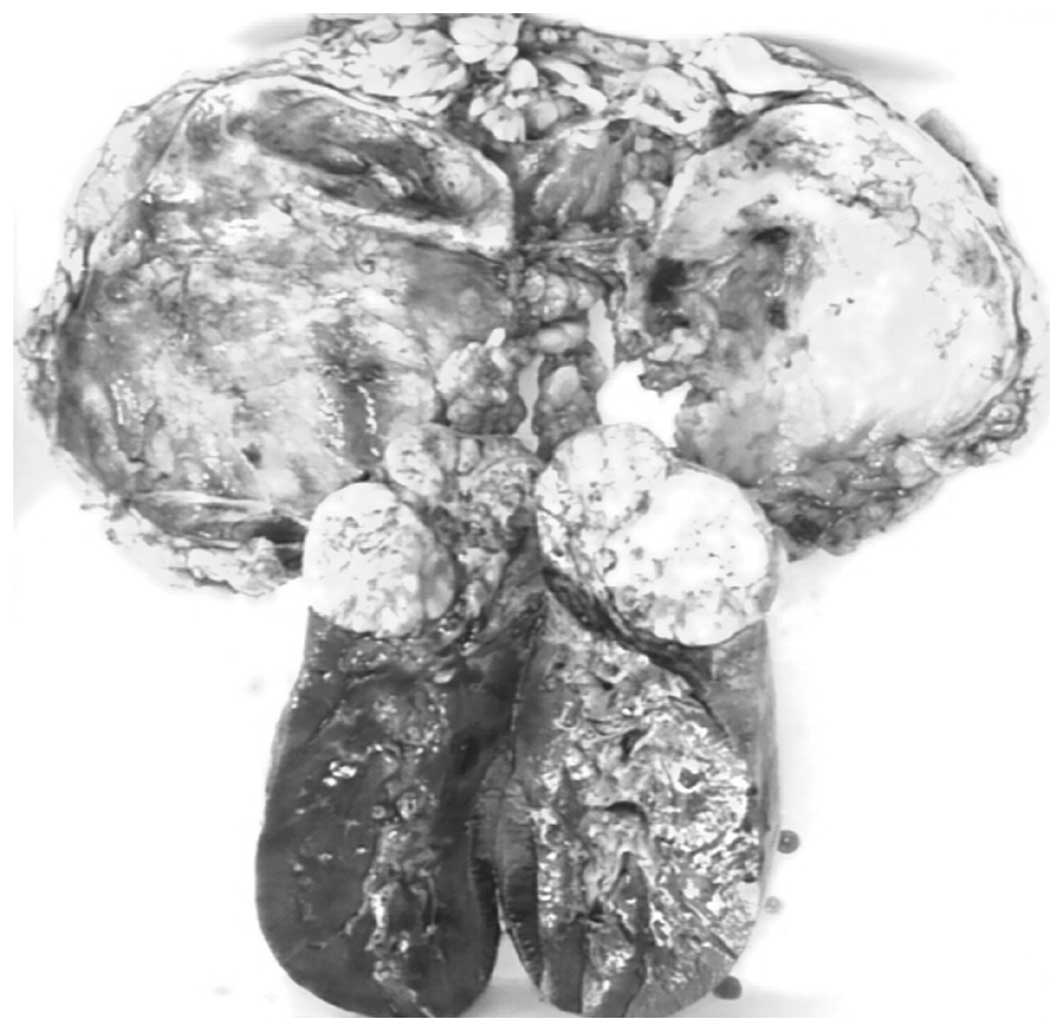
radical nephrectomy. Surgery revealed one mass in the lower pole of
the kidney which extended locally into the capsule. The cut surface
of the tumor was white to gray with a fleshy texture and focal
areas of necrosis and hemorrhage (Fig.
2).
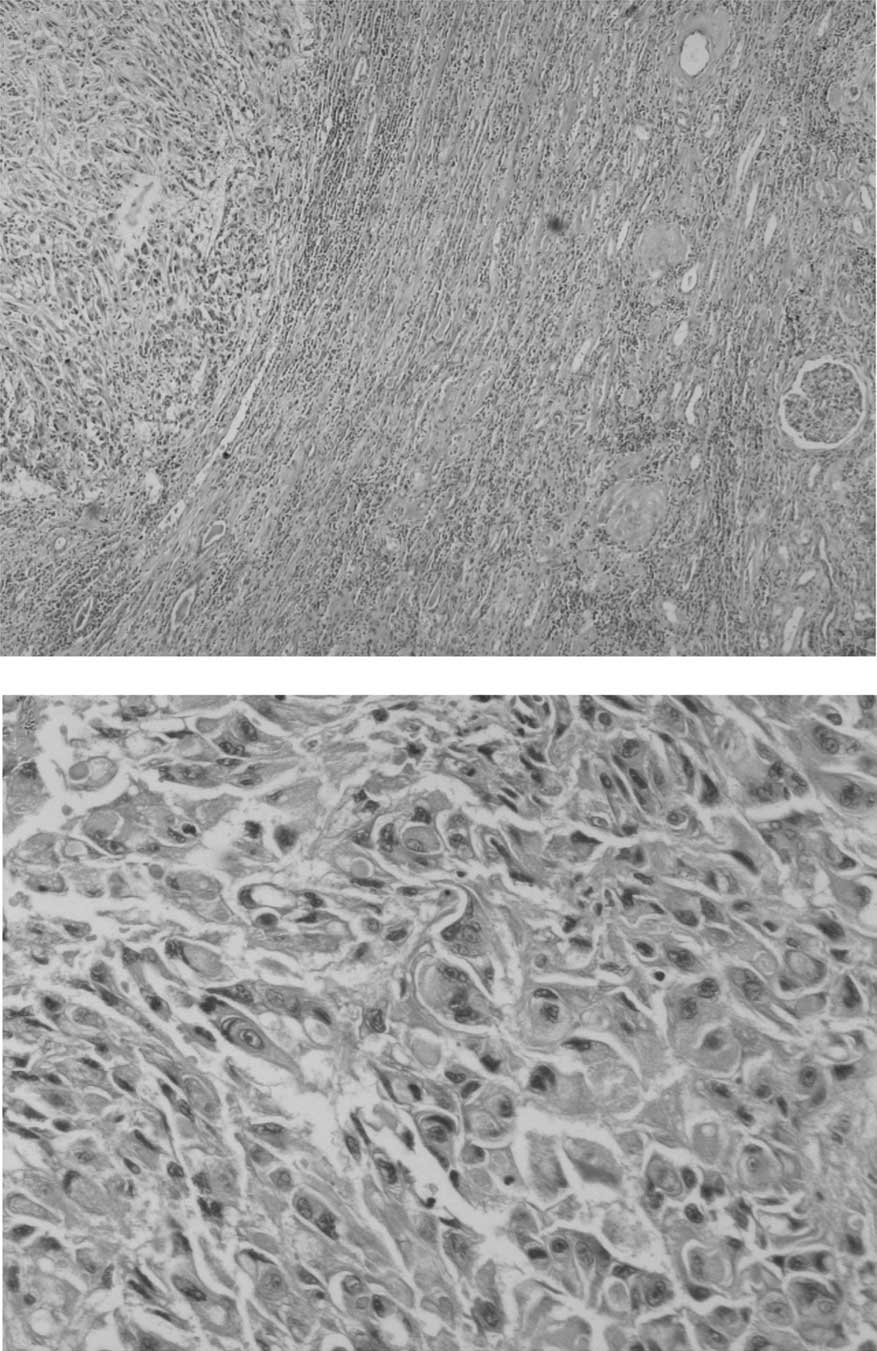
Microscopically, the tumor cells were noncohesive,
large and round to polygonal, with vesicular nuclei and prominent
nucleoli. A number of the neoplastic cells had eccentric nuclei and
large round eosinophilic cytoplasmic inclusion-like structures.
Certain areas of the tumor exhibited hemorrhage and/or necrosis
(Fig. 3A and B).
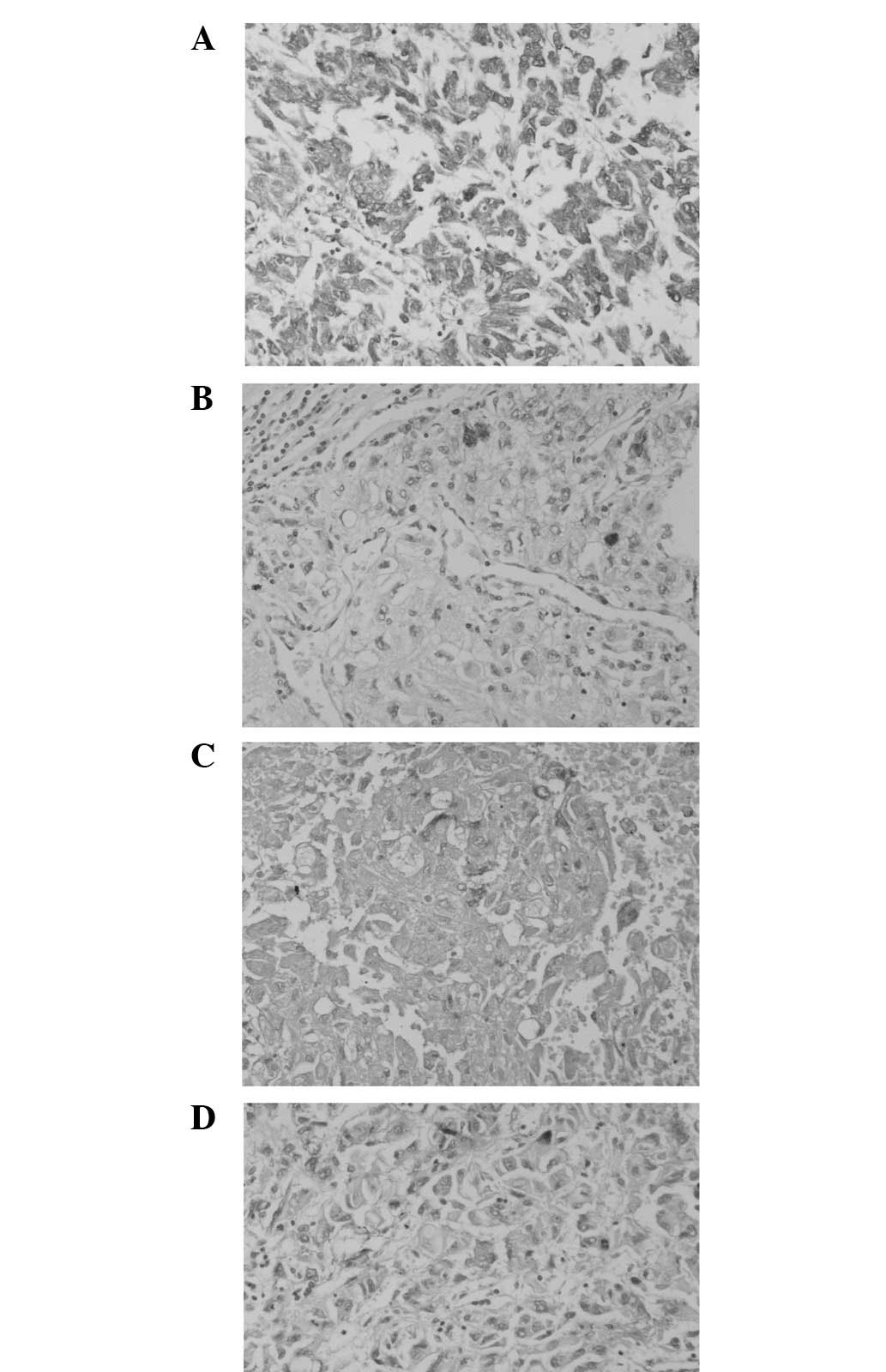
Immunohistochemically, the tumor cells were diffusely positive for
vimentin (Fig. 4A) and focally
positive (cytoplasmic and dotlike) for neuron-specific enolase
(NSE; Fig. 4B), S-100 (Fig. 4C) and epithelial membrane antigen
(EMA; Fig. 4D). The staining was
negative for pan-cytokeratin, CK7, myoglobin, desmin,
muscle-specific actin (MSA) and smooth muscle actin (SMA).
Accordingly, a pathological diagnosis of pure
malignant rhabdoid tumor of the left kidney was established based
on the microscopic features and immunohistochemical findings.
A 10-month postoperative follow-up revealed no
indications of tumor recurrence or metastasis.
Discussion
MRTKs were originally described as a
‘rhabdomyosarcomatoid variant of Wilm’s tumor’ by Beckwith and
Palmer in 1978 due to the resemblance of the cells to
rhabdomyoblasts (6). Subsequently,
this type of tumor was recognized as a distinct and unique
malignant renal tumor. MRTK has also been described at extrarenal
sites in children and adults, including the urogenital system such
as the bladder or prostate (7–9).
MRTK may be associated with other malignancies
(composite) or constitute the only component (pure). Several
carcinomas arising in the kidney have been observed to have
rhabdoid features, including renal clear cell, papillary,
chromophobe, transitional cell and collecting duct carcinomas
(10–12). However, only five cases of pure
adult MRTK have been reported in the English-language literature.
The present study describes the sixth case of pure adult MRTK.
Patients typically exhibit an abdominal mass but
only a minority have hematuria and flank or abdominal pain. In a
small subset of patients the tumor is discovered incidentally in
radiological examination (1–5).
However, haematuria is a common symptom in children. Symptoms
arising from metastases are common at diagnosis since metastases
occur in ∼80% of patients, mainly affecting the lungs, liver and
brain. Patients may also experience reversible hypercalcaemia as a
result of increased parathyroid hormone concentration (13). The characteristic findings of
imaging studies are often not present in adult patients, whereas CT
findings suggesting rhabdold tumors of kidney, including
calcification, subcapsular hematoma and the lobular appearance of a
large, centrally located heterogeneous renal mass, are often
observed in children (14). In the
present case, the patient did not have the perceived clinical
symptoms and no characteristics were observed in the imaging
studies. Thus, the asymptomatic mass may have been be at an early
stage.
Macroscopically, MRTK is generally a single, poorly
circumscribed mass, located in the middle of the kidney which
exhibits infiltrating growth. Sections of MRTKs are white to
grayish with fleshy or soft textures. Necrosis and hemorrhage foci
may also be observed. The classic histological appearance of MRTK
is that of patternless sheets of noncohesive, large, ovoid or round
to polygonal cells with vesicular nuclei and prominent nucleoli,
with certain neoplastic cells exhibiting eccentric nuclei and large
round eosinophilic cytoplasmic inclusion-like structures. These
were all consistent with the present case. In addition, certain
inflammatory cells, particularly lymphocytes or histiocytes, may
accompany the neoplastic cells and the rhabdoid elements are of
high nuclear grade. Ultrastructurally, the rhabdoid cells have
paranuclear intermediate filament aggregates, which are arranged
haphazardly. In addition, paranuclear condensation of organelles
associated with peripheral vacuolization also occurs (1–5,15,16).
Immunohistochemically, the tumor cells are
frequently stained diffusely positive for vimentin and focally
positive for EMA, cytokeratin, NSE and S-100. However, staining for
myoglobin, desmin, MSA, SMA, human melanoma, black-45 (HMB-45) and
CD99 are often negative (1–5,15,16),
which is consistent with the present case.
Molecular studies have revealed an apparently
characteristic presence of mutations in the hSNF5/INI1 gene on
chromosome 22, which has become the hallmark of cranial, renal and
other rhabdoid tumors, particularly in children. The lack of
immunohistochemical staining for the INI1 gene product facilitates
the diagnosis of such diseases. In adults, the rhabdoid phenotype
is viewed as a non-specific morphological feature due to the
‘dedifferentiation’ common to numerous neoplasms, including various
carcinomas, sarcomas and meningiomas. These tumors do not exhibit
the deletion of the h5NF5/INI1 gene so the nuclear immunostaining
for INI1 is retained. However, it remains possible to identify the
inactivation of h5NF5/INI1 in certain adult MRTs when properly
tested. Consequently, whether all tumors within one individual
exhibit the downregulation of hSNF5/INI1 remains controversial
(17).
In the present case, although electron microscopic
examination and genetic and/or molecular analysis were not
performed due to technical limitations at the time, a pathological
diagnosis of pure MRTK was established based on the histological
and immunohistochemical analysis, whereas several previous
researchers have based the diagnosis of rhabdoid tumors solely on
morphological and immunohistochemical findings (1–3).
Surgery is considered to be the first choice of
treatment if possible. The former postoperative treatment for MRTK
cases in children was radiotherapy or a four-drug regimen for high
risk nephroblastoma patients (18).
However, considering the dismal outcome of full chemotherapy or
radiotherapy, more effective treatments such as molecular-targeted
or neoadjuvant therapy approaches are highly desirable. Kapoor
et al’s study reported a response to sorafenib with overall
disease stabilization in a patient with adult rhabdoid RCC,
indicating that tyrosine kinase inhibitors may be used for
effective therapeutic strategies (19). Koos et al noted that cell
lines obtained from rhabdoid tumors of the kidney and extrarenal
rhabdoid tumors consistently expressed the tyrosine kinase c-Abl.
Treatment with the tyrosine kinase inhibitor imatinib, resulted in
reduced cellular growth in the two cell lines. This study
demonstrated a functional role for c-Abl in the biology of rhabdoid
tumors and provided a rationale for the investigation of tyrosine
kinase inhibitors that target c-Abl for the treatment of these
tumors (20). Venneti et al
observed that MRTs expressed numerous stem cell-associated
transcription factors, which may be regulated by the expression of
EZH2 and the Id family of proteins. This study demonstrated
similarities between MRTs and stem cells and it may aid in the
elucidation of common biological pathways that could serve in
advancing more effective therapeutic strategies for treating MRTs
(21). These studies all indicate
that a deeper understanding of the biology of these aggressive
tumors is likely to aid in the design of more effective
therapies.
The prognosis is generally poor for MRTKs and the
majority recur and/or metastasize. Studies indicate that there are
no differences in survival due to the primary site, gender, or
ethnicity in children. The stage of the tumor is negatively
correlated with survival, while the use of radiotherapy is
positively associated with survival. In addition, patients younger
than 2 or older than 18 years at diagnosis have lower survival
rates than patients between 2 and 18 years. Adult patients have
improved outcomes compared with young children (<2 years old)
but a poorer outcome compared with older children (2–18 years old).
The tumor stage significantly affects the outcome but the use of
radiotherapy is not associated with the difference in outcome
(17). In the present case, tumor
recurrence or metastasis were not observed at the 10-month
postoperative follow-up. However, long term follow-up is essential
since metastases may occur after several years in view of the
malignancy.
In conclusion, the present study reports one case of
pure adult MRTK with characteristic clinicopathological features.
Considering the fact that the characteristic findings are often not
observed in clinical symptom and imaging studies, the histological
features, immunohistochemical staining and cytogenetic studies may
aid in confirming the diagnosis of pure MRTK. In addition, further
study and a larger case series are required to fully clarify the
natural history and identify the ideal postoperative treatment
protocol for these aggressive tumors.
References
|
1
|
Lowe W, Weiss RM, Todd MB and True LD:
Malignant rhabdoid tumor of the kidney in an adult. J Urol.
143:110–112. 1990.PubMed/NCBI
|
|
2
|
Clausen HV, Horn T, Anagnostaki L and
Larsen S: Malignant rhabdoid tumour of the kidney in an adult: a
case report with immunohistochemical and ultrastructural
investigation. Scand J Urol Nephrol Suppl. 157:123–128.
1994.PubMed/NCBI
|
|
3
|
Ebbinghaus SW, Herrera G and Marshall ME:
Rhabdoid tumor of the kidney in an adult: review of the literature
and report of a case responding to interleukin-2. Cancer Biother.
10:237–241. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caballero JM, Collera P, Marti L, et al:
Malignant rhabdoid tumor of the kidney in the adult. Actas Urol
Esp. 20:377–379. 1996.(In Spanish).
|
|
5
|
Peng HQ, Stanek AE, Teichberg S, et al:
Malignant rhabdoid tumor of the kidney in an adult: a case report
and review of the literature. Arch Pathol Lab Med. 127:e371–e373.
2003.PubMed/NCBI
|
|
6
|
Beckwith JB and Palmer NF: Histopathology
and prognosis of Wilms’ tumor results from the first National
Wilms’ Tumor Study. Cancer. 41:1937–1948. 1978.
|
|
7
|
Palmer NF and Sutow W: Clinical aspects of
the rhabdoid tumor of the kidney: a report of the National Wilms’
Tumor Study Group. Med Pediatr Oncol. 11:242–245. 1983.PubMed/NCBI
|
|
8
|
Savage N, Linn D, McDonough C, et al:
Molecularly confirmed primary malignant rhabdoid tumor of the
urinary bladder: implications of accurate diagnosis. Ann Diagn
Pathol. 16:504–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim JY, Cho YM and Ro JY: Prostatic
stromal sarcoma with rhabdoid features. Ann Diagn Pathol.
14:453–456. 2010. View Article : Google Scholar
|
|
10
|
Kumar S, Kumar D and Cowan DF:
Transitional cell carcinoma with rhabdoid features. Am J Surg
Pathol. 16:515–521. 1992. View Article : Google Scholar
|
|
11
|
Weeks DA, Beckwith JB, Mierau GW and
Zuppan CW: Renal neoplasms mimicking rhabdoid tumor of kidney. A
report from the National Wilms’ Tumor Study Pathology Center. Am J
Surg Pathol. 15:1042–1054. 1991.PubMed/NCBI
|
|
12
|
Yusuf Y, Belmonte AH and Tchertkoff V:
Fine needle aspiration cytology of a recurrent malignant tumor of
the kidney with rhabdoid fetures in an adult. A case report. Acta
Cytol. 40:1313–1316. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahmed HU, Arya M, Levitt G, et al: Part I:
Primary malignant non-Wilms’ renal tumours in children. Lancet
Oncol. 8:730–737. 2007.
|
|
14
|
Chung CJ, Lorenzo R, Rayder S, et al:
Rhabdoid tumor of the kidney in children: CT findings. AJR Am J
Roentgenol. 164:697–700. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gökden N, Nappi O, Swanson PE, et al:
Renal cell carcinoma with rhabdoid features. Am J Surg Pathol.
24:1329–1338. 2000.
|
|
16
|
Shannon B, Stan Wisniewski Z, Bentel J and
Cohen RJ: Adult rhabdoid renal cell carcinoma. Arch Pathol Lab Med.
126:1506–1510. 2002.PubMed/NCBI
|
|
17
|
Sultan I, Qaddoumi I, Rodríguez-Galindo C,
et al: Age, stage, and radiotherapy, but not primary tumor site,
affects the outcome of patients with malignant rhabdoid tumors.
Pediatr Blood Cancer. 54:35–40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van den Heuvel-Eibrink MM, van Tinteren H,
Rehorst H, et al: Malignant rhabdoid tumours of the kidney (MRTKs),
registered on recent SIOP protocols from 1993 to 2005: a report of
the SIOP renal tumour study group. Pediatr Blood Cancer.
56:733–737. 2011.PubMed/NCBI
|
|
19
|
Kapoor A, Tutino R, Kanaroglou A and Hotte
SJ: Treatment of adult rhabdoid renal cell carcinoma with
sorafenib. Can Urol Assoc J. 2:631–634. 2008.PubMed/NCBI
|
|
20
|
Koos B, Jeibmann A, Lünenbürger H, et al:
The tyrosine kinase c-Abl promotes proliferation and is expressed
in atypical teratoid and malignant rhabdoid tumors. Cancer.
116:5075–5081. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Venneti S, Le P, Martinez D, et al:
Malignant rhabdoid tumors express stem cell factors, which relate
to the expression of EZH2 and Id proteins. Am J Surg Pathol.
35:1463–1472. 2011. View Article : Google Scholar : PubMed/NCBI
|