Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare
disease entity that has been reported to occur in multiple
anatomical locations, including the lung, bladder, spleen, breast,
pancreas, liver, colon, prostate, peripheral nerves, soft tissue
and orbit (1). IMT has thus far
been most commonly referred to in the literature as ‘inflammatory
pseudotumor’, ‘plasma cell granuloma’, ‘pseudosarcoma’ or
‘fibromyxoid lesion’.
Histologically, these lesions may exhibit a
heterogeneous appearance, thus accounting for the various terms
used to describe IMTs alluded to above. All IMTs are characterized
by variable cellular spindle cell proliferation with a compact
myxoid stromal pattern and a variable inflammatory infiltrate,
usually comprised of plasma cells, lymphocytes, eosinophils and
neutrophils. The spindle cells possess the morphological appearance
of myofibroblasts. Immunohistochemistry of the spindle cells
reveals reactivity for smooth muscle actin and desmin, and
ultrastructural studies reveal a predominance of myofibroblasts and
a smaller fibroblastic component (1).
IMTs may occur in any age group, however, they are
observed most commonly in children and adolescents. The most
prevalent anatomical sites for IMTs to occur include lung,
abdomino-pelvic and retroperitoneal areas, although any site may be
involved. Patients may present with non-specific symptoms
associated with the site of the mass, including cough and abdominal
pain. Constitutional symptoms are present in 15–30% of cases and
laboratory evaluation may reveal microcytic anemia, an elevated
ESR, thrombocytosis and/or polyclonal hypergammaglobulinemia
(1). In some cases, the mass may be
detected after an extensive work-up of fever of unknown origin.
These systemic symptoms frequently resolve following surgical
excision and tumor recurrence may be marked by a return of clinical
and laboratory abnormalities.
IMTs are classified as tumors with an intermediate
biological potential, in that local recurrences may occur and there
is a rare possibility of distant metastasis (2,3).
Previously, insight into the pathogenesis of at
least some cases of IMT has been illuminated by the finding of
rearrangements of the anaplastic lymphoma kinase (ALK) gene on
chromosome 2p23 in 50% of IMT cases. Multiple fusion partners have
been identified. ALK expression by immunohistochemistry reliably
predicts a rearrangement by FISH or PCR (1).
Complete surgical resection, whenever feasible, is
the treatment of choice for IMT (4,5).
Ill-circumscribed tumors, particularly in the abdomino-pelvic area,
may be difficult to completely resect and local recurrences are not
uncommon. Local recurrences are usually managed with re-excision if
possible and the vast majority of patients with local recurrence
are free of disease with long-term follow-up (4,5).
For patients who are unable to have complete
resections, including in the case of multiple lesions or disease in
an area where resection would be anatomically difficult, other
modalities have been employed. Corticosteroid monotherapy may
result in rapid resolution of the disease and sustained remission
(6,7). Non-steroidal anti-inflammatory agents
(NSAIDs) as solitary therapy may be extremely efficacious (8) and are the subject of this study.
Radiation alone may induce enduring remission (9). Anecdotal response to chemotherapy has
also been reported (10).
Most recently, a case has been published in which
crizotinib, an inhibitor of ALK kinase, induced a partial remission
in a patient with an IMT characterized by a RANB2-ALK fusion gene
(11). A patient in the same study
with an ALK-negative IMT did not respond to crizotinib, supporting
ALK inhibition as the basis of the therapeutic effect (11).
We report our experience in the successful treatment
of an ALK-negative IMT of the lung, pre-treated with two surgeries
and corticosteroid therapy, with a prolonged course of celecoxib
and have comprehensively reviewed the literature on NSAID therapy
of IMTs. Informed consent was obtained from the patient.
Case report
Clinical presentation and diagnosis
A 52-year-old woman was observed to have a pulmonary
nodule in the left lower lobe on a chest X-ray, performed as part
of preoperative testing for a dilatation and curettage. Computed
tomography (CT) scans of the chest performed subsequently showed
nodular opacities in the bilateral lower lobes. The patient was
managed conservatively and a repeat CT scan performed 2 months
thereafter showed almost complete resolution of the nodule in the
left lower lobe and complete resolution of the nodule in the right
lower lobe. At the time, these findings were thought to represent
the resolution of inflammatory disease of the lung.
In October 2007, viral-type respiratory symptoms led
to a CT scan of the chest which revealed a left lower lobe lung
nodule. PET/CT scans in November 2007 revealed a 2.3×1.9-cm
irregularly shaped nodule in the left lower lobe with minimal
uptake (SUV 3.4) as well as a separate 7-mm non-avid nodule.
Follow-up chest CT scans in December 2007 revealed that the left
lower lobe nodule resolved into two separate nodules, in aggregates
larger than the previous. Multiple new pulmonary nodules were
observed in both lungs. A wedge resection of the left lower lobe in
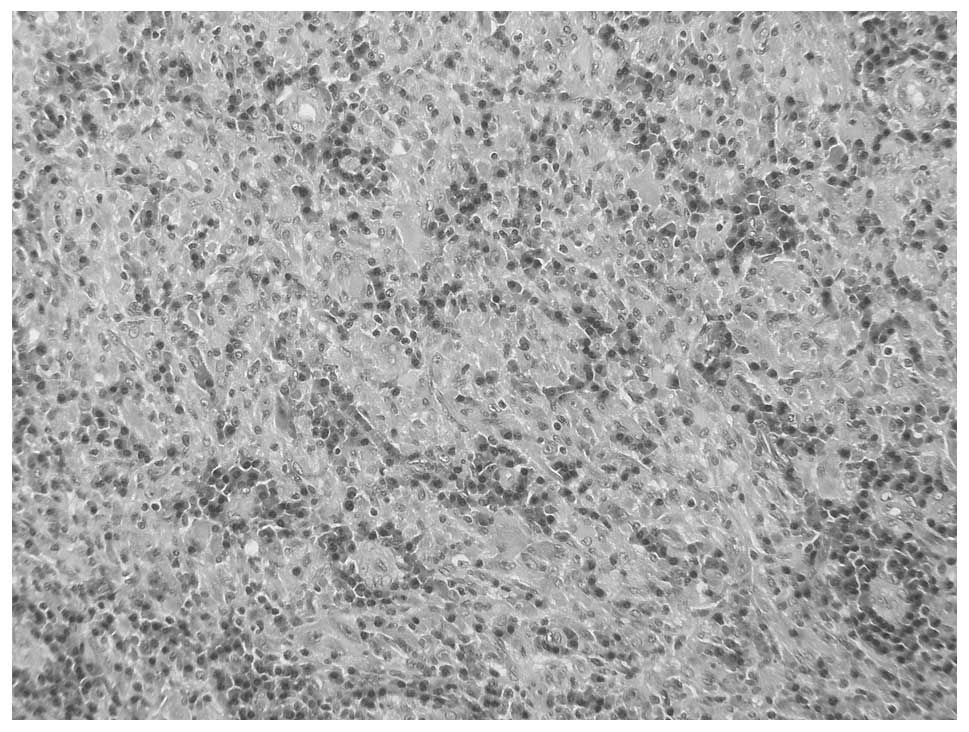
January 2008 revealed a mass composed of plasma cells admixed with
lymphocytes, histiocytes and mesenchymal cells (Fig. 1). Review at the Pathology Department
of the National Institutes of Health (Bethesda, MD, USA) confirmed
the diagnosis of plasma cell granuloma. We have since performed ALK
immunohistochemistry on this specimen, which identified no
positivity.
Treatment and clinical course
Follow-up CT scans of the chest were performed
thereafter. In March 2009, there was progression of disease with
new nodules compared with an assessment performed in December 2008.
In April 2009, repeat CT showed increases in size of the left upper
lobe and left lingular nodules. The patient underwent surgical
resection of the left lingular mass in May 2009, again revealing
plasma cell granuloma. Repeat scans performed one month after the
second surgery demonstrated progression with multiple nodules
involving bilateral lung fields. This prompted treatment with
prednisone, initiated in June 2009 at 40 mg/day, which was reduced
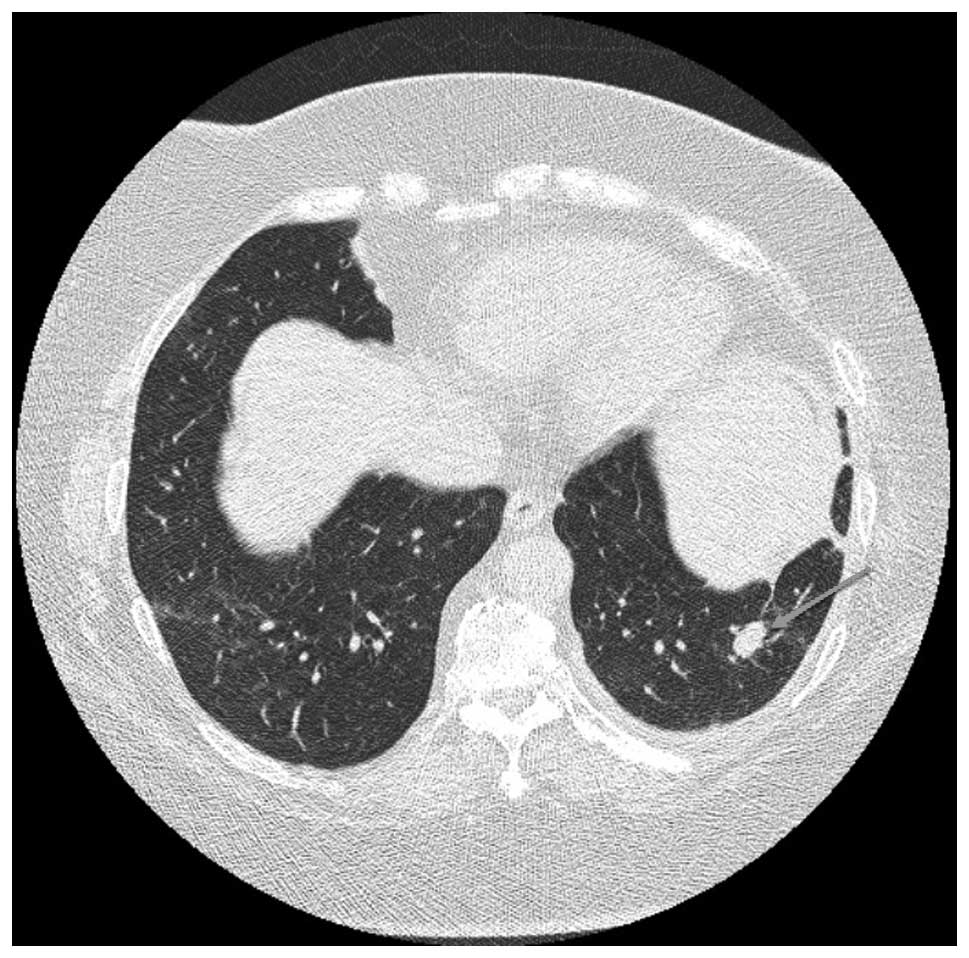
by October 2009. CT scans in September had revealed resolution of
the nodules, however, in December 2009, a follow-up scan
highlighted the recurrence of multiple lung nodules involving
bilateral lung fields (Fig. 2). In
January 2010, the patient was administered celecoxib 200 mg PO BID.
By March 2010, CT scans showed improvement with resolution of a
nodule and a reduction in size of others, with subsequent scans
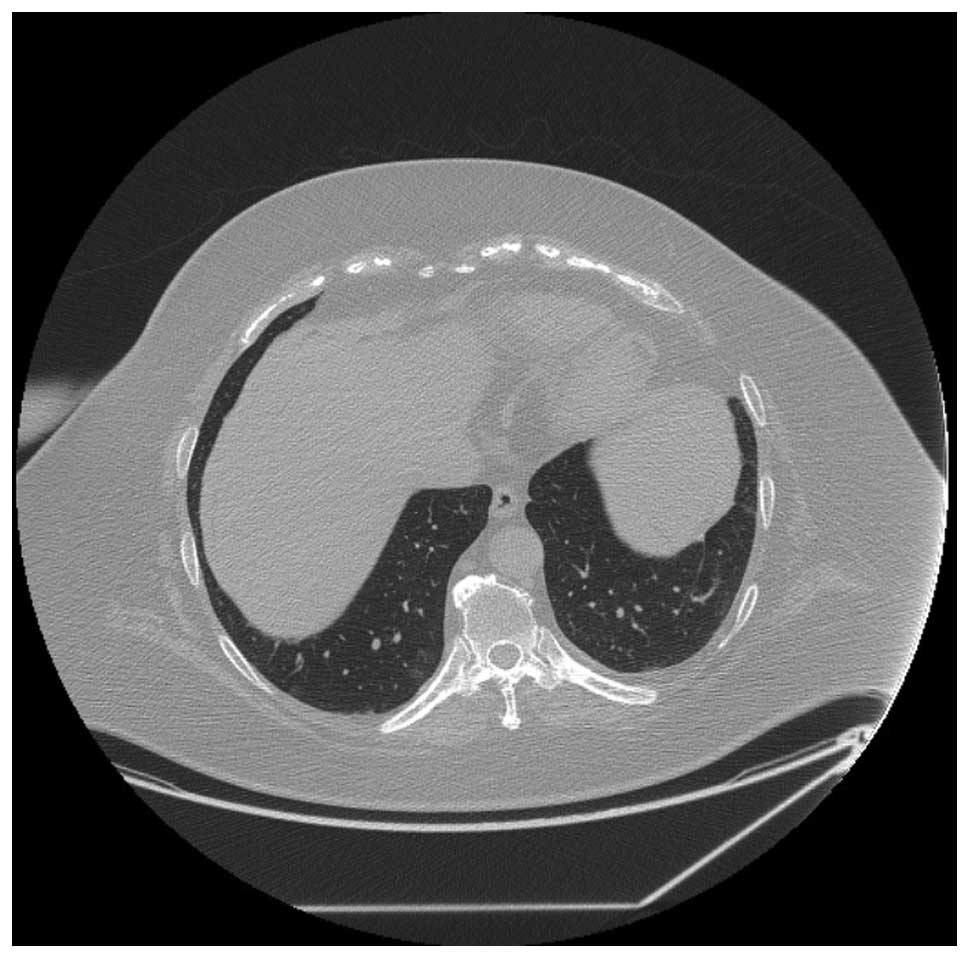
revealing continuing response to therapy. By February 2012, CT
scans demonstrated resolution of all lung nodules (Fig. 3). The patient’s most recent CT scan
in August 2012 revealed no progression of disease, 32 months after
starting celecoxib. The patient has been reduced to 200 mg every
other day.
Literature review search strategy
A literature search was performed in MEDLINE and
EMBASE to identify all published studies using the search terms
‘inflammatory myoblastic tumor’, ‘plasma cell granuloma’,
‘inflammatory pseudotumor’, ‘NSAIDs’, ‘anti-inflammatory agents,
nonsteroidal’ and ‘cyclooxgenase 2 inhibitors’. References for all
retrieved studies were also reviewed to ensure that no studies had
been missed in the primary search.
Summary of retrieved studies
Altogether, there have been a total of eight
previous studies of IMTs managed successfully with NSAID
monotherapy, comprising a total of ten patients (Table I). In the first reported case,
Hakozaki et al(8) treated a
female with a 2.5×2.5-cm tumor in the left lobe of the liver. The
biopsy revealed ‘edematous granulation tissue with capillaries and
fibroblasts’. The patient had experienced abdominal pain for 6
months; one month of non-steroidal therapy induced complete
remission (CR) of the mass and resolution of the abdominal
discomfort. Su et al(12)
reported on two patients with intra-abdominal masses. Tissue
pathology was not described other than being consistent with an
inflammatory pseudotumor (IPT). NSAID monotherapy induced CR after
2 months in one patient and after 4 months in the other patient. In
both cases, NSAID therapy was discontinued at the time of achieving
CR and the patients subsequently remained in remission. The case
report of Chan et al(13) is
the only case of NSAID therapy of pulmonary IMT other than our own
case. The reported patient had an unresectable right lung mass, was
febrile and toxic-appearing, with laboratory results indicative of
acute inflammation with thrombocytosis, elevated ESR and elevated
CRP. Histology revealed fibrous tissue, areas of necrosis and
extensive lymphocyte and plasma cell infiltration. Rofecoxib
therapy caused marked symptomatic improvement within 72 h;
inflammatory parameters normalized and the patient achieved CR
after 8 months of therapy. Przkora et al(14) published two cases of intra-abdominal
IMT. Both patients received a short initial period of steroid
treatment along with an NSAID, which was subsequently continued as
monotherapy. The response trajectory is not described in either
patient. One patient achieved CR after 14 months and the other had
stable disease after 12 months. Both patients were maintained on
NSAIDs at the time of publication. Colakoglu et al(15) reported on a case of a female with a
solitary liver mass. NSAID therapy induced CR by 40 days.
Vassiliadis et al(16) also
reported on a patient with a solitary liver mass. In this case, the
patient had fever, leucocytosis thrombocytosis and an acute phase
reaction serology, i.e., the same clinical presentation as the case
of Chan et al(13). Naproxen
induced a prompt reduction in temperature and was continued for one
month. Off therapy, the mass continued to regress and the patient
achieved CR. Mattei and Barnaby (17) reported a case in which ketorolac
induced a significant response in a large retroperitoneal mass; the
patient required surgery within 24 h due to rapid regression of the
tumor resulting in a perforated duodenum. Finally, Colangelo et
al(18) reported a case of a
pancreatic head IMT in which ibuprofen induced CR after 6 months.
Four years after ceasing drug administration, the patient remained
in an unmaintained remission.
 | Table IPublished studies of NSAID therapy of
IMT. |
Table I
Published studies of NSAID therapy of
IMT.
| Author (Ref.) | Age (years) | Gender | Primary site | Prior treatment | Therapy
(duration) | Result
(duration) |
|---|
| Present case | 52 | F | Lung | Two surgeries
Corticosteroids | Celecoxib (32
months) | CR (32 months) |
| Colangelo et
al(18) | 4 | M | Pancreas | None | Ibuprofen (6
months) | CR (4 years) |
| Mattei et
al(17) | 13 | M | Duodenum | None | Ketorolac (24 h) | CR (NS) |
| Vassiliadis et
al(16) | 16 | M | Liver | None | Naproxen (1
month) | CR (1 year) |
| Colakoglu et
al(15) | 62 | F | Liver | None | NSAID (1 month) | CR (1 year) |
| Przkora et
al(14) | 63 | F | Mesentery | None | Diclofenac
(ongoing) | CR (14 months) |
| 22 | M | Retroperitoneum | None | Ibuprofen
(ongoing) | SD (1 year) |
| Chan et
al(13) | 7 | F | Lung | None | Rofecoxib (8
months) | CR (NS) |
| Su et
al(12) | 6 | F | Pelvis | None | Naproxen (4
months) | CR (2 years) |
| 14 | M | Mesentery | None | Ibuprofen (2
months) | CR (6 months) |
| Hakozaki et
al(8) | 52 | F | Liver | None | Loxoprofen (1
month) | CR (6 months) |
Thus, of the ten cases of NSAID therapy reported in
the literature, nine were in CR and one had stable disease at the
time of publication. The marked paucity of studies in the
literature search is noteworthy, and likely reflects a publication
bias, in that a study of unsuccessful use of NSAID therapy would
not have been submitted for publication.
From these studies, it is apparent that i) responses
are not site-specific; ii) responses occur extremely soon after the
onset of treatment, indeed as early as 24 h; iii) CRs to NSAID
therapy may be extremely durable; iv) remission persists after
termination of NSAID therapy.
Discussion
The induction of durable CR of IMTs with NSAID
therapy, albeit a likely rare phenomenon given the scarcity of
published studies, prompts speculation as to the mechanism of
antitumor activity in this setting.
Cyclooxygenase (COX) is the key enzyme involved in
the synthesis of important biological modulators called
prostanoids, a collective term for prostaglandins and thromboxanes.
Prostanoids are involved in multiple physiological processes,
including vasomotility, platelet aggregation, gastrointestinal
mucosal integrity, immunomodulation and regulation of cell growth
and differentiation (19). There
are two major isoforms of COX; COX-1 which is constitutively
expressed in all normal tissues and COX-2, which is an enzyme that
is inducible by inflammatory and mitogenic signals.
COX-2 products have been demonstrated to be
important in cancer development, progression and metastasis
(19,20). Prostaglandin E2 (PGE2 ) is
particularly significant in mediating tumor progression.
Overexpression of COX-2 in tumors leads to progression via multiple
mechanisms, including increasing angiogenesis, decreasing apoptosis
and increasing invasiveness. PGE2 initiates a downstream growth
signaling cascade involving the epidermal growth factor receptor,
nuclear receptor ras-mitogen-activated protein kinase pathways and
upregulating invasiveness by activating matrix
metalloproteinases.
Numerous types of cancer have been demonstrated to
over-express COX-2 and overexpression often correlates with more
aggressive behaviour (20). Given
the importance of COX-2 in enhancing cell growth, COX-2 inhibitors
have been employed in a number of clinical settings (19,20).
The most notable data have been in the area of chemoprevention,
particularly in the reduction of the development of colonic polyps.
Despite a large body of work demonstrating a significant antitumor
effect in preclinical models, results have been disappointing in
the advanced cancer setting thus far.
Applebaum et al studied the expression of
COX-2, VEGF and ALK in 11 cases of IMT (21). Using a grading system of 1+ to 3+,
with 3+ representing the greatest staining intensity, one tumor was
graded as 3+, seven as 2+ and one as 1+. VEGF was strongly
expressed in these tumors, suggesting an association of COX-2 with
increased angiogenesis. On the basis of these data, the authors
postulated that COX-2 inhibition may be an effective therapy for
IMT. IMT additionally likely includes a spectrum of disease and
based on anecdotal data, abdominal tumors may respond more
favorably than thoracic tumors to COX inhibition (21).
NSAID therapy may induce enduring CR in untreated or
pretreated IMT, as evidenced by our case and the abovementioned
studies. Our patient had recurrent disease after surgery,
suggesting a more aggressive biology, and corticosteroid therapy
only achieved a transient response (6 months). Significantly, our
case also demonstrates that NSAID therapy may induce CR in
ALK-negative IMTs where ALK inhibition is not a therapeutic option.
Excellent response to an NSAID is likely a rare event, however,
given the fact that responses may be marked and persist after
ceasing therapy, a trial of an NSAID should be considered in any
patient with an unresectable or recurrent IMT.
Acknowledgements
The authors thank Janice Lester for
her assistance with the literature search.
References
|
1
|
Gleason BC and Hornick JL: Inflammatory
myofibroblastic tumours: where are we now? J Clin Pathol.
61:428–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Coffin CM and Fletcher JA: Inflammatory
myofibroblastic tumor. Fletcher CD, Unni KK and Mertens F; World
Health Organization Classification of Tumors: Pathology and
Genetics of Soft Tissue and Bone. IARC Press; Lyon: pp. 91–93.
2002
|
|
3
|
Coffin CM, Hornick JL and Fletcher CD;
Inflammatory myoblastic tumor: Comparison of clinicopathologic,
histologic, and immunohistochemical features including ALK
expression in atypical and aggressive cases. Am J Surg Pathol.
31:509–520. 2007.PubMed/NCBI
|
|
4
|
Kovach SJ, Fischer AC, Katzman PJ, Salloum
RM, Ettinghausen SE, Madeb R and Koniaris LG: Inflammatory
myofibroblastic tumors. J Surg Oncol. 94:385–391. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coffin CM, Watterson J, Priest JR and
Dehner LP: Extrapulmonary inflammatory myofibroblastic tumor
(inflammatory pseudotumor). A clinicopathologic and
immunohistochemical study of 84 cases. Am J Surg Pathol.
19:859–872. 1995. View Article : Google Scholar
|
|
6
|
Lee MH, Lee HB, Lee YC, Rhee YK, Lee EJ,
Chung MJ, Jin GY, Kweon EY and Park SJ: Bilateral multiple
inflammatory myofibroblastic tumors of the lung successfully
treated with corticosteroids. Lung. 189:433–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carswell C and Chataway J: The successful
long-term management of an intracranial inflammatory myoblastic
tumor with corticosteroids. Clin Neurol Neurosurg. 114:77–79. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hakozaki Y, Katou M, Nakagawa K, Shirahama
T and Matsumoto T: Improvement of inflammatory pseudotumor of the
liver after nonsteroidal anti-inflammatory agent therapy. Am J
Gastroenterol. 88:1121–1122. 1993.PubMed/NCBI
|
|
9
|
Imperato JP, Folkman J, Sagerman RH and
Cassady JR: Treatment of plasma cell granuloma of the lung with
radiation therapy. A report of two cases and review of the
literature. Cancer. 57:2127–2129. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bertocchini A, Lo Zupone C, Callea F,
Gennari F, Serra A, Monti L and de Ville de Goyet J: Unresectable
multifocal omental and peritoneal inflammatory myofibroblastic
tumor in a child: revisiting the role of adjuvant therapy. J
Pediatr Surg. 46:e17–e21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Butrynski JE, D’Adamo DR, Hornick JL, Dal
Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ,
Ramaiya N, Kwak EL, Clark JW, Wilner KD, Christensen JG, Jänne PA,
Maki RG, Demetri GD and Shapiro GI: Crizotinib in ALK-rearranged
inflammatory myofibroblastic tumor. New Engl J Med. 363:1727–1733.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Su W, Ko A, O’Connell TX and Applebaum H:
J Pediatr Surg. 35:1635–1637. 2000. View Article : Google Scholar
|
|
13
|
Chan PW, Omar KZ and Ramanujam TM:
Successful treatment of unresectable inflammatory pseudotumor of
the lung with COX-2 inhibitor. Pediatr Pulmonol. 36:167–169. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Przkora R, Bolder U, Schwartz S, Jauch KW,
Spes J, Andreeson R and Mackensen A: Regression of nonresectable
inflammatory myofibroblastic tumours after treatment with
nonsteroidal anti-inflammatory drugs. Eur J Clin Invest.
34:320–321. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Colakoglu O, Unsal B, Haciyanli M, Tunakan
M, Buyrac Z, Yorukoglu G, Yazicioglu N and Genc H: A successfully
managed inflammatory pseudotumor of liver without surgery: report
of a case. Acta Gastroenterol Belg. 68:382–384. 2005.PubMed/NCBI
|
|
16
|
Vassiliadis T, Vougiouklis N, Patsiaoura
K, Mpoumponaris A, Nikolaidis N, Giouleme O and Evgenidis N:
Inflammatory pseudotumor of the liver successfully treated with
nonsteroidal anti-inflammatory drugs: a challenge diagnosis for one
not so rare entity. Eur J Gastroenterol Hepatol. 19:1016–1020.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mattei P and Barnaby K: Rapid regression
of duodenal inflammatory myofibroblastic tumor after intravenous
ketorolac: case report and review of the literature. J Pediatr
Surg. 43:1196–1199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Colangelo M, Lisi G and Chiesa PL:
Pancreatic inflammatory myofibroblastic tumor (IMT). J Pediatr
Surg. 45:1074–1075. 2009.PubMed/NCBI
|
|
19
|
Liao Z, Mason K and Milas L:
Cyclo-oxygenase-2 and its inhibition in cancer. Drugs. 67:821–846.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masferrer JL, Leahy KM, Koki AT, Zweifel
BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ and
Seibert K: Antiangiogenic and antitumor activities of
cyclooxygenase-2 inhibitors. Cancer Res. 60:1306–1311.
2000.PubMed/NCBI
|
|
21
|
Applebaum H, Kieran MW, Cripe TP, Coffin
C, Collins MH, Kaipainen A, Laforme A and Shamberger RC: The
rationale for nonsteroidal anti-inflammatory drug therapy for
inflammatory myofibroblastic tumors: a Children’s Oncology Group
Study. J Pediatr Surg. 40:999–1003. 2005.PubMed/NCBI
|