Introduction
The homeostatic balance of reactive oxygen species
(ROS) and redox reactions is critically significant for the
maintenance of cellular physiological processes. However, an
imbalance of the cellular oxygen environment leads to oxidative
cellular stress resulting in deleterious consequences, including
DNA damage and the activation of pro-survival signaling molecules
(i.e., NFκB and MAPK) that contribute to the etiology and
progression of human cancers (1–3). The
adverse effects of unregulated ROS levels on human cancers have
recently gained considerable attention as therapeutic targets for
the treatment of this disease (4–6). Thus,
the present study examined the anti-tumor properties of the
antioxidant 1′-acetoxychavicol acetate (ACA), a ginger-derived
natural product extract from the rhizomes and seeds of Alpinia
galanga, on brain tumors.
Mechanistically, ACA acts as an inhibitor of
xanthine oxidase, which plays a significant role in the catabolism
of purines, and catalyzes the oxidation of hypoxanthine to
xanthine, and xanthine to uric acid. Additionally, xanthine oxidase
plays a significant role in the generation of several ROS,
including H2O2, O2− and
OH−(7). Byproducts of
xanthine oxidase oxidation have been implicated in several abnormal
physiological processes, including brain ischemia, vascular injury
and inflammatory diseases (7). Of
particular significance for the present study is the expression of
xanthine oxidase expressed at increased levels in brain tumors
compared to normal brain tissue (8). In contrast, several scavenging
antioxidant enzymes have been shown to be decreased in meningiomas,
astrocytomas and medulloblastomas (9). Furthermore, although early studies by
Ohnishi et al(10) and
Tanaka et al(11)
demonstrated that the xanthine oxidase inhibitor, ACA, was a
chemopreventive agent that suppressed tumor formation in the oral
cavity and colon of rats, subsequent studies revealed that ACA
exerted positive anti-tumorigenic effects on leukemia and breast
and multiple myeloma cancers (12–14).
The present study demonstrates that ACA antagonizes glioblastoma
cell migration and proliferation as a consequence of caspase
3-activated cell death.
Materials and methods
Cells conditions and reagents
U373, U87 and A172 glioblastoma cells were purchased
from the American Type Culture Collection (Manassas, VA, USA). The
study was approved by the Ethics Committee of the Southern
University at New Orleans (New Orleans, LA, USA). All cell lines
were maintained in Dulbecco’s modified Eagle’s medium (DMEM)
(Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum
(Invitrogen), 2 mM L-glutamine (Invitrogen), 100 nM minimal
essential medium (MEM) non-essential amino acids (Invitrogen),
penicillin (5000 units/ml) and streptomycin (5000 μg/ml)
(Invitrogen) at 37°C in 5% CO2.
Crystal violet cell proliferation
assay
The cells were plated in 12-well plates, treated
with 10, 5 and 2 μM ACA and allowed to incubate for 48 h
(vehicle controls were treated with dimethyl sulfoxide; DMSO).
Next, the tissue culture medium was removed; the cell monolayer was
fixed with 100% methanol for 5 min and stained with 0.5% crystal
violet in 25% methanol for 10 min. The cells were then washed with
distilled water 3 times for 5 min each to remove any excess dye and
allowed to dry overnight at room temperature. The incorporated dye
was then solubilized in 0.1 M sodium citrate (Sigma-Aldrich, St.
Louis, MO, USA) in 50% ethanol. Next, 100 μl of the treated
and control samples were transferred to 96-well plates and the
optical densities were read at 540 nm using an X-mark microplate
absorbance spectrophotometer (BioRad, Hercules, CA, USA).
Clonogenic survival
The cells were plated for 24 h, treated with 1
μM ACA or DMSO (vehicle) and allowed to incubate at 37°C for
10–14 days. At the termination of the incubation period, the cells
were fixed with absolute methanol, stained with 1% crystal violet
for 10 min, rinsed in tap water and allowed to dry. Colonies,
consisting of ≥50 cells, were then counted to determine the
survival fraction.
Cell motility
Cell motility assays were conducted using
polycarbonate membrane inserts of CytoSelect Cell Migration Assays
according to the manufacturer’s instructions (Cell Biolabs Inc.,
San Diego, CA, USA). A cell suspension containing
0.5–1.0×106 cells/ml was prepared in serum-free media
with vehicle (DMSO) or 5 μM ACA, while 500 μl of
media containing 10% fetal bovine serum was added to the lower
chamber of the migration plate. A total of 300 μl of the
cell suspension containing vehicle or 5 μM ACA was then
added to the inside of each plate insert and allowed to incubate
for 24 h at 37°C in 5% CO2. Subsequently, the
non-migratory cells were removed from the plate inserts (per the
manufacturer’s instructions) and the migratory cells were
counterstained according to manufacturer’s instructions (Cell
Biolabs Inc.).
Cell adhesion assay
The cell suspensions containing
0.1–1.0×106 cells/ml in serum-free media with vehicle
(DMSO) or 5 μM ACA, were plated onto collagen IV adhesion
plates for use in a CytoSelect Cell Adhesion Assay (Cell Biolabs
Inc.) for 24 h. Subsequently, the media containing vehicle (DMSO)
or 5 μM ACA was removed and the cells were stained and
solubilized according to the manufacturer’s instructions (Cell
Biolabs Inc.). Optical density measurements (540 nm) were then
collected for the cell extracts to quantitate the adhesive
properties of the cells treated with vehicle DMSO or 5 μM
ACA.
Caspase 3 activity
The cells were plated in serum-free DMEM for 24 h,
treated with 2 μM ACA or vehicle (DMSO) and allowed to
incubate for 24 h. The cells were then lysed in CelLytic M Cell
Lysis reagent (Sigma-Aldrich) and the protein concentrations were
determined using the Bradford method. Subsequently, caspase 3
activity assays were conducted with a CaspACE Assay System
according to the manufacturer’s instructions (Promega, Madison, WI,
USA) using 30 μg protein.
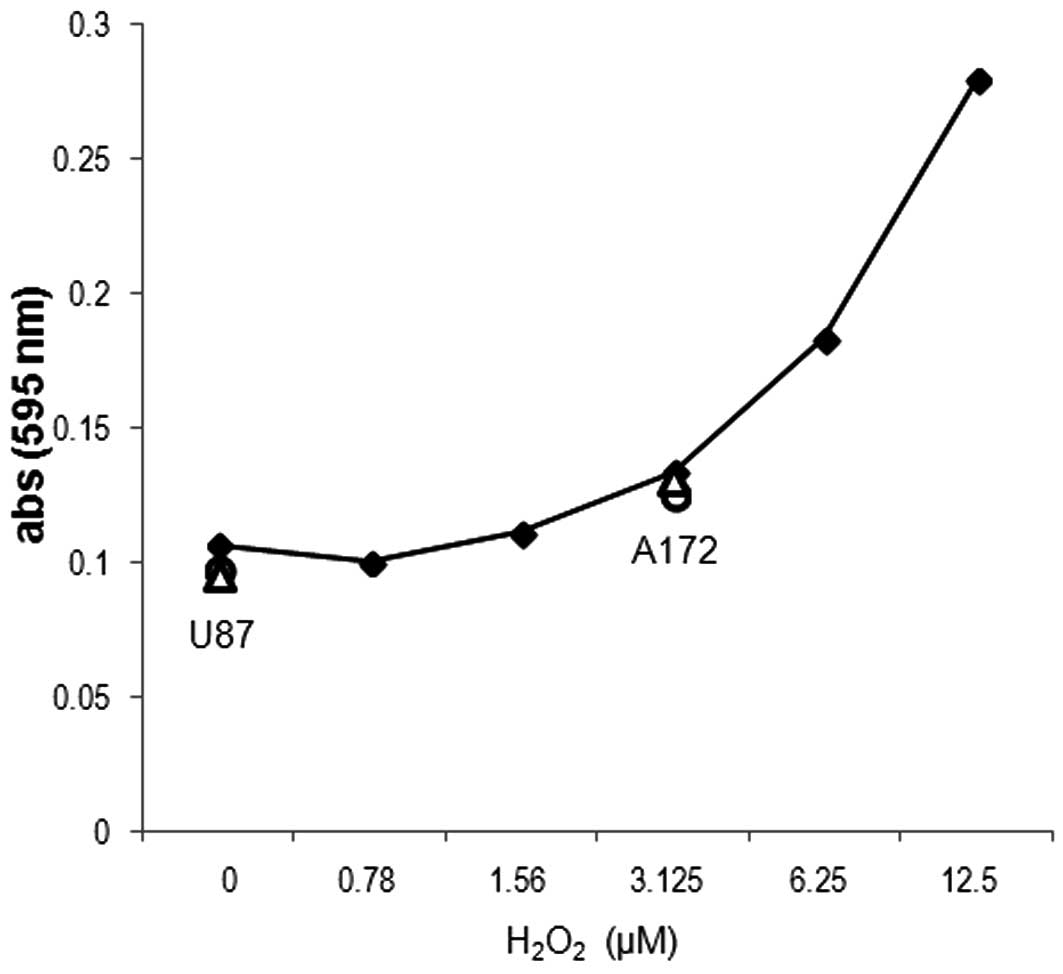
ROS (H2O2)
analysis
ROS assays were conducted using an OxiSelect
Hydrogen Peroxide Assay in accordance with the manufacturer’s
instructions (Cell Biolabs Inc.). A serial dilution of
H2O2 (0–100 μM) was prepared to
generate a standard curve, while U87 and A172 cells
(1×107) were plated, exposed to 5 μM ACA or
vehicle (DMSO) for 24 h and then sonicated. Subsequently, standards
and samples were mixed with an aqueous working reagent (per the
manufacturer’s instructions), incubated on a shaker for 30 min at
room temperature and absorbances were read using an ELx808
Absorbance Microplate Reader (BioTek, Winooski, VT, USA) at 595
nm.
Cytokine array assays
U87 and A172 cells were plated overnight, treated
with 5 μM ACA or vehicle (DMSO) for 2 h and lysed with
CelLytic M Cell Lysis reagent. The protein concentrations were
determined using the Bradford method. Cytokine array assays were
conducted using a Human 4-Plex Cytokine Array according to the
manufacturer’s instructions (Quansys Biosciences, Logan, UT, USA).
A serial dilution was performed with a positive control standard
antigen in order to generate variable cytokine expression. Diluted
standards and 20 μg of protein lysis extract from the
treated and vehicle controls were added to wells containing 4-plex
arrays (IL-1α, IL-4, IL-6, TNFα and a reference spot) and incubated
on a plate shaker for 1 h at room temperature. The wells were then
washed 3 times with washing buffer, a detection mix was added and
the wells were again incubated on a plate shaker for 1 hour at room
temperature. Next, the wells were washed 3 times,
streptavidin-horseradish peroxidase (HRP) was added for 15 min,
followed by 6 washes, and a substrate for the detection of the
cytokines was added. Subsequently, the multiplex plate arrays were
imaged using a Q-View imager (Quansys Biosciences).
Results
ACA inhibits glioblastoma cell
proliferation and migration
In the present study, the effects of the xanthine
oxidase inhibitor, ACA, on glioblastoma cells were initially
evaluated with dose-response experiments, which displayed an
overall decrease in cell proliferation in response to increasing
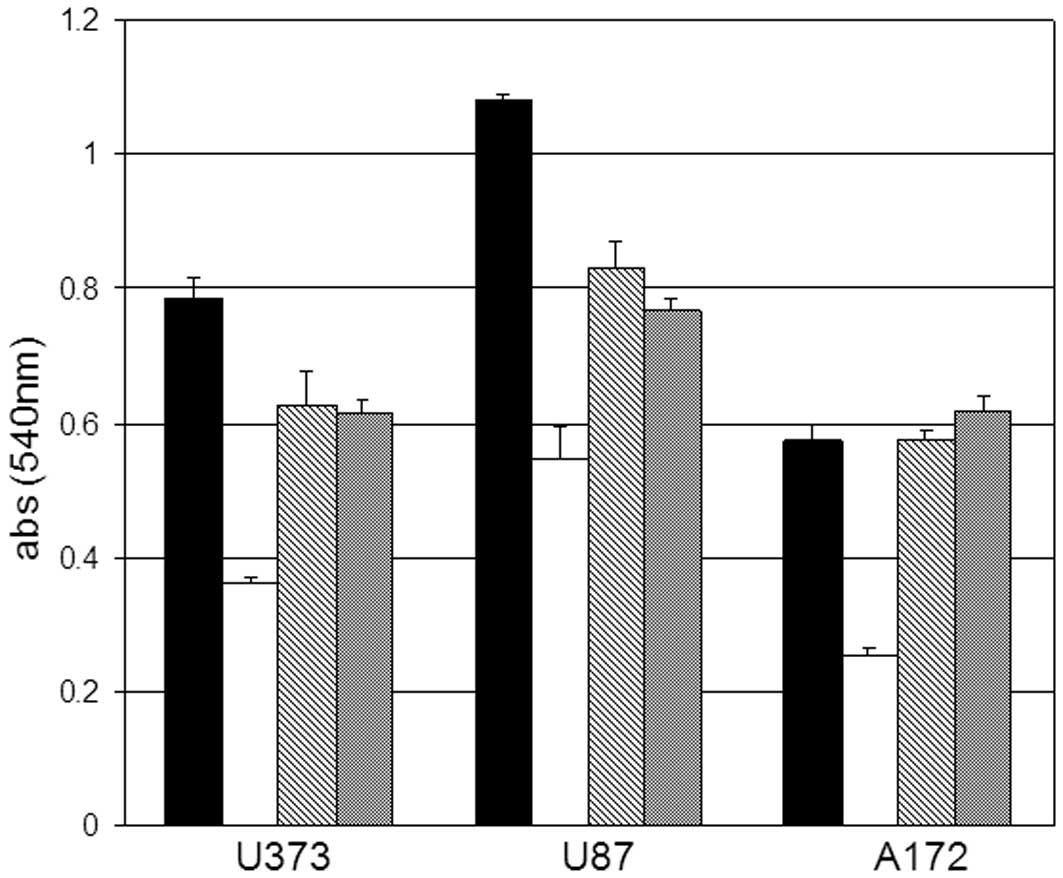
concentrations of ACA 48 h post-exposure. Glioblastoma cells
treated with 10 μM ACA displayed the most significant
inhibition on cell proliferation compared to the vehicle-treated
control cells (Fig. 1). U373 and
U87 cell proliferation was also impaired by exposure to 5 μM
and 2 μM ACA (Fig. 1) in
contrast to the A172 cells treated with the same concentrations of
ACA. To further assess the cytotoxic effects of ACA on glioblastoma
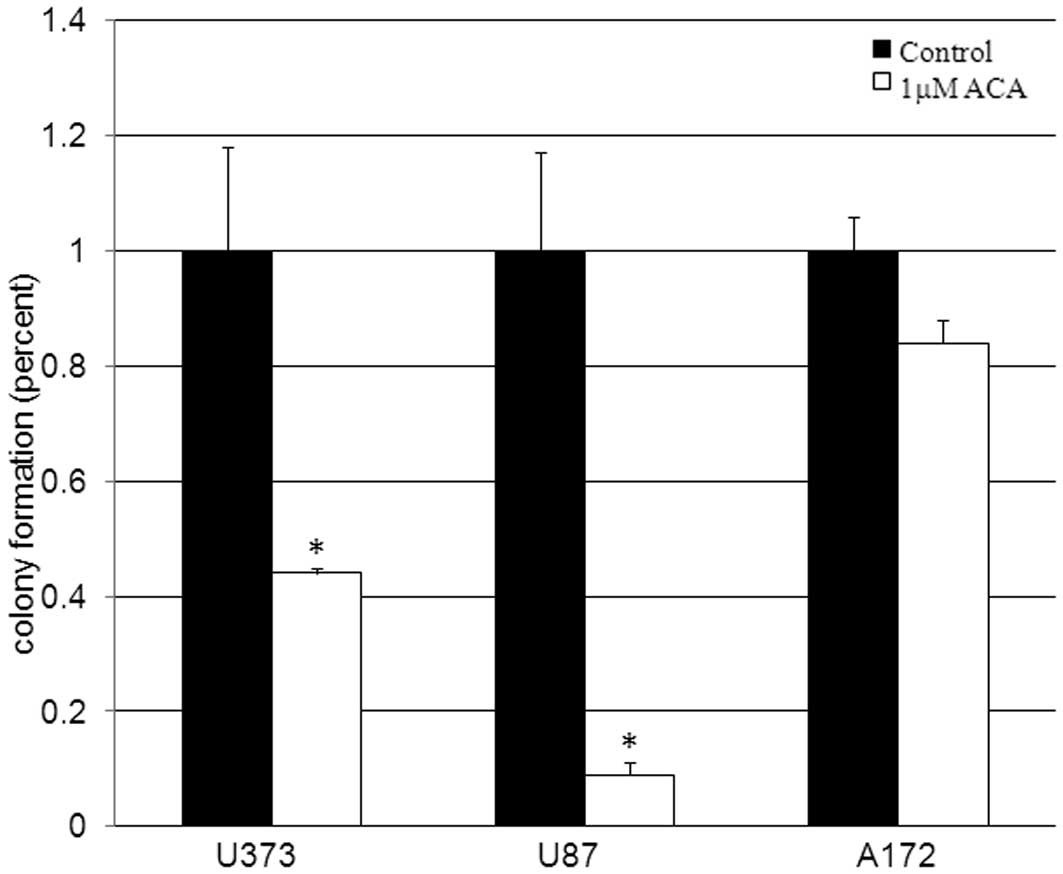
cells clonogenic survival assays were conducted. Clonogenic
survival assays utilize low cell plating densities (<500 cells)
to evaluate the ability of single cells to form colonies, making it
a sensitive assay that assesses cellular reproductive capacity and
mimics the clonal expansion behavior of human cancers. Clonogenic
survival data revealed statistically significant (P<0.05)
decreases of 56 and 91% in the cellular reproductive capacity of
the U373 and U87 cells, respectively, when treated with 1 μM
ACA (Fig. 2) compared with the
vehicle-treated control cells. These data are consistent with
previous studies on leukemia (12)
and breast cancer (14) cells that
also demonstrated the anti-proliferative effects of ACA on these
human cancers.
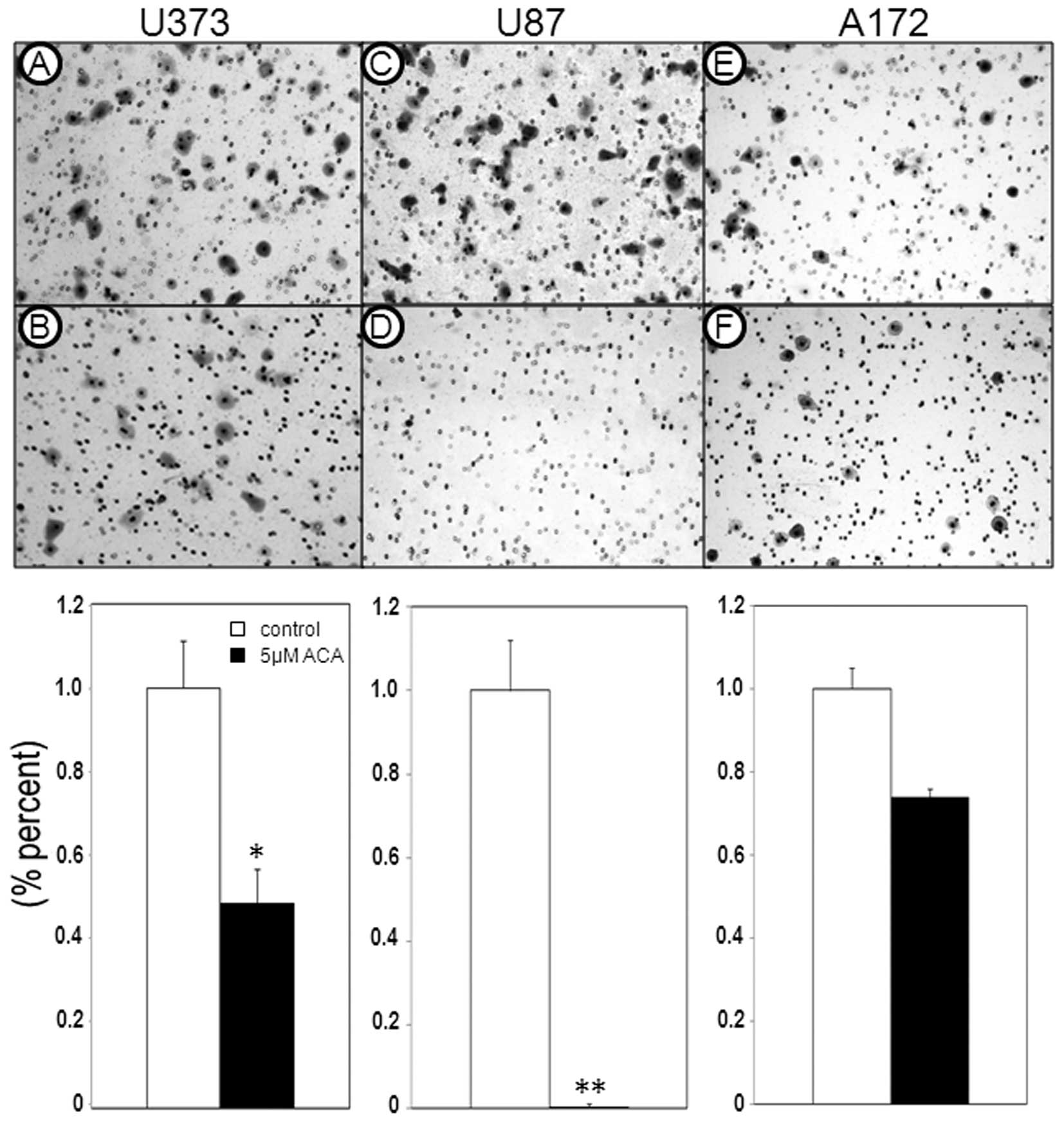
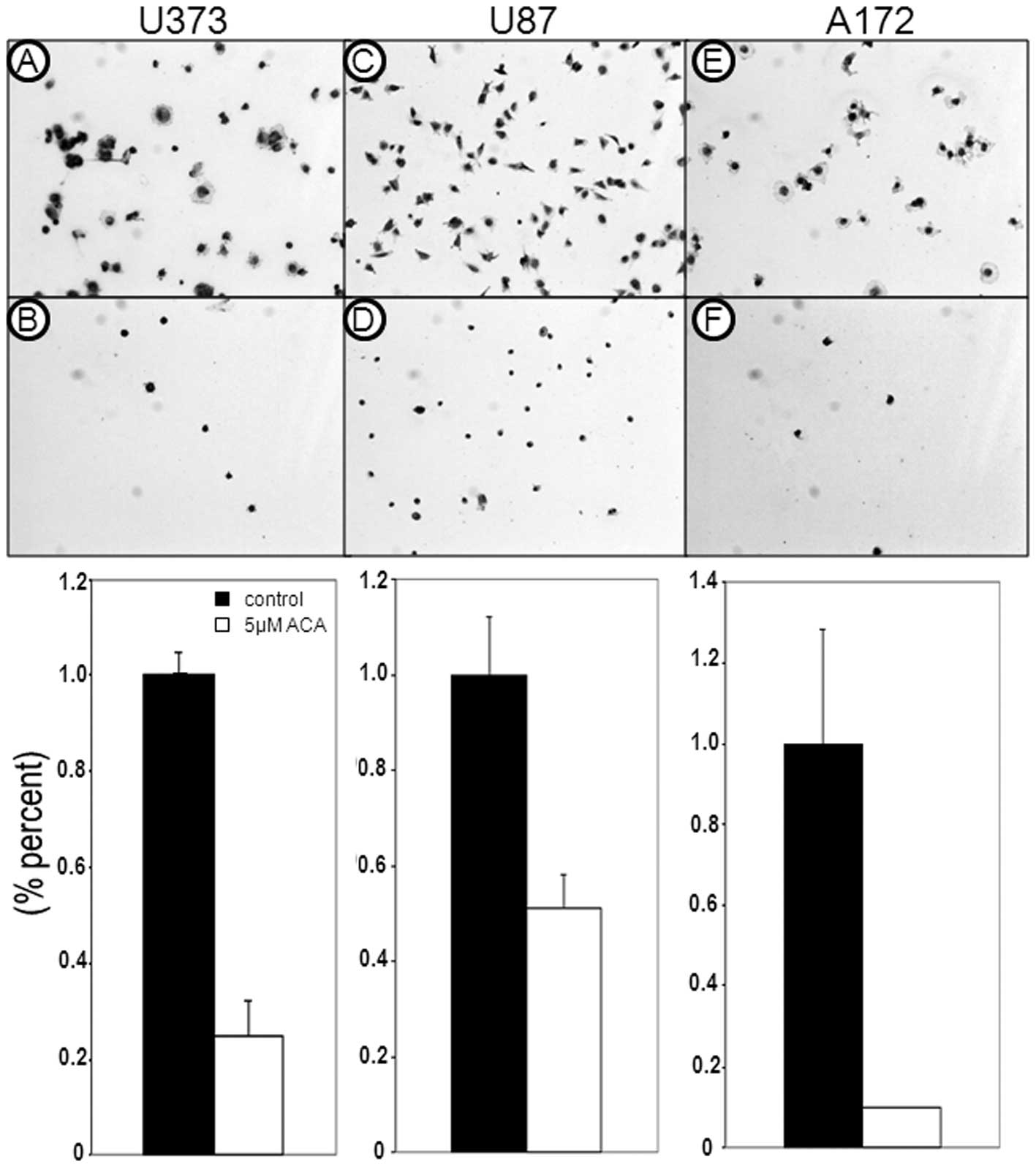
To date, little is known regarding the efficacy of
xanthine oxidase inhibitors on cell migration, particularly tumor
cell migration. Therefore, in addition to examining the effects of
ACA on glioblastoma cell proliferation, the ability of ACA to
antagonize glioblastoma cell migration was also assessed. Migration
assays revealed a reduction in the migratory ability of the
glioblastoma cells following 5 μM ACA exposure (Fig. 3); the most significant effect was
observed in the U87 cells, which showed a 97% reduction in cell
migration (P<0.01), followed by the U373 cells, which displayed
a 51% reduction in cell migration (P<0.05) compared with the
vehicle-treated control cells. In addition, ACA also caused a
reduction in glioblastoma cell adhesion (Fig. 4), paralleling the observed decrease
in glioblastoma cell migration post-ACA treatment.
Caspase 3-activated cell death
The cytotoxic and apoptotic-inducing ability of
Alpinia galanga rhizome extract and ACA have been
demonstrated in several types of human cancers (12–19).
Therefore, by evaluating caspase 3 activity, the present study
examined whether the observed inhibitory effects of ACA on
glioblastoma cell production were a consequence of apoptotic cell
death. Caspase 3, an effector (executioner) caspase of apoptotic
cell death, was measured in the glioblastoma cells treated with 2
μM ACA. Increases in caspase 3 activity of 23 and 49% were
observed in the U87 and A172 glioblastoma cells, respectively,
compared with the vehicle-treated control cells (Fig. 5). This data provides evidence that
indicates that glioblastoma cells undergo apoptosis in response to
ACA exposure and is consistent with a study by Moffatt et
al, which demonstrated that ACA promoted apoptotic cell death
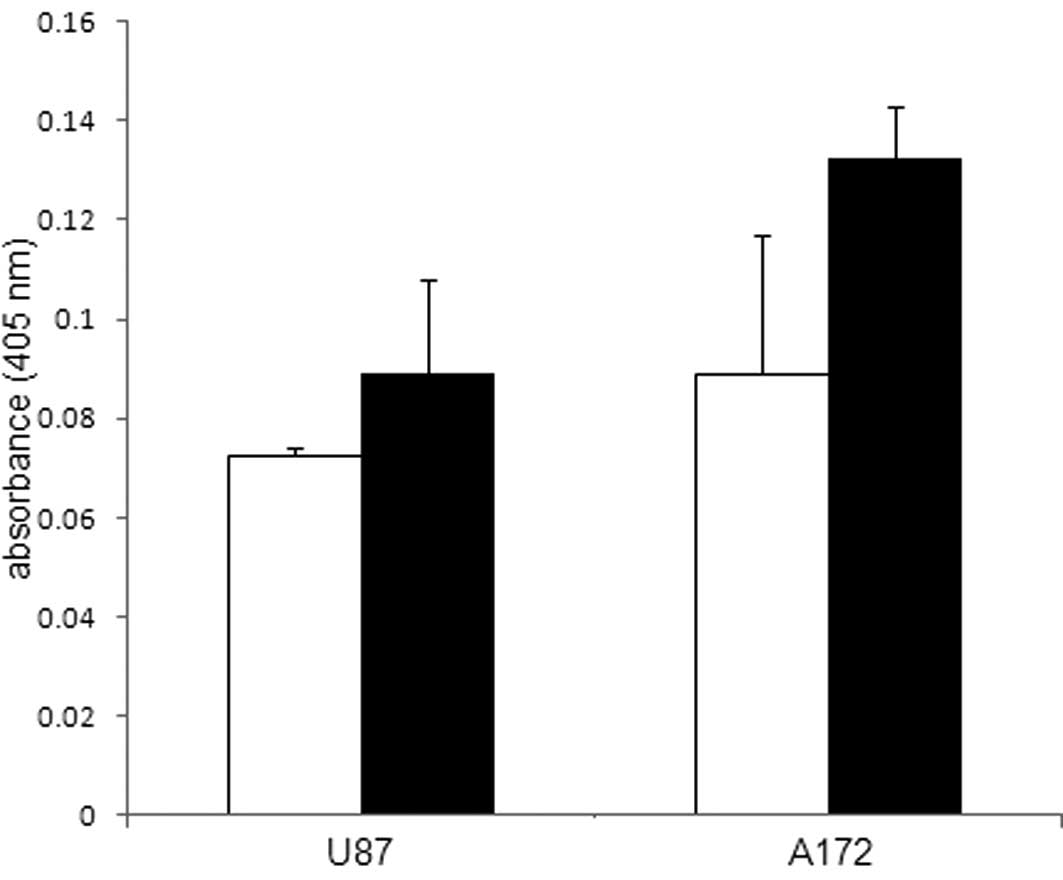
in Ehrlich ascites tumor cells via caspase 3 activation (15). In addition to assessing caspase 3
involvement during ACA-induced glioblastoma cell death, ROS, which
are known to influence tumor cell viability were also studied in
the ACA-treated glioblastoma cells. An evaluation of the ROS levels
was performed by measuring the H2O2
concentration; however, no notable difference was observed between
glioblastoma cells exposed to 5 μM ACA and the
vehicle-treated control cells (Fig.
6).
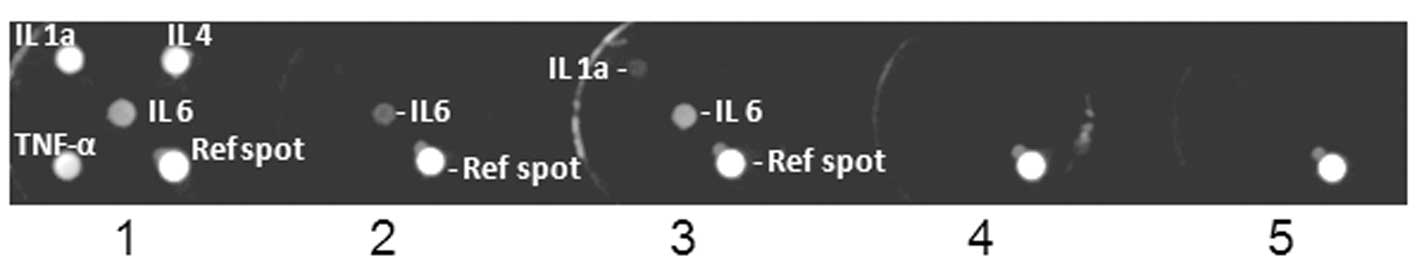
ACA-induced cytokine expression
Cytokines are well-recognized as molecules that
contribute to the development, progression and maintenance of
several types of human cancers as a consequence of pleiotropic
signaling mechanisms that promote the survival of tumor cells
(20,21). Recent studies have demonstrated that
ROS promote cytokine production (22) and provide mechanistic support for
cytokine activity. The present study therefore evaluated the effect
of ACA on cytokine expression in glioblastomas (Fig. 7), which are known to express IL-6 at
high levels compared with normal brain tissue, and have been shown
to play a prominent role in glioblastoma cell survival and
migration (23–26). A cytokine expression array analysis
revealed that the treatment with 2 μM ACA invoked the
increased expression of IL-6 and IL-1α in the U87 cells compared
with the vehicle-treated controls, while cytokines were not
detected in the A172 cells (Fig.
7).
Discussion
Antioxidants are often studied for their properties
as chemopreventive agents. However, in the present study, the
utility of the antioxidant, ACA, was examined for its
anti-tumorigenic properties on glioblastomas. Glioblastomas, like
normal brain tissue, have low antioxidant activity. This is
supported by several studies that have revealed depleted levels of
ROS detoxification enzymes (gluthathione peroxidase and superoxide
dismutase) in clinical glioblastomas examined from tumor patient
explants (27–29). Additionally, glioblastomas have been
shown to increase the expression levels of the ROS generator,
xanthine oxidase. Together, these studies suggest that ROS, which
have been shown to have pro-tumorigenic effects, contribute to the
survival, maintenance and progression of glioblastomas.
The present study demonstrated that ACA antagonizes
glioblastoma cell proliferation as a consequence of promoting
caspase 3-induced apoptotic cell death. These findings parallel
results previously shown in myeloma (13), leukemia (12) and breast cancer studies (14), which also demonstrated that ACA
invoked an apoptotic cell death response as a result of increased
caspase 3 activity. In contrast to the well-characterized
inhibitory effects of ACA on tumor cell proliferation, few studies
have been conducted on the efficacy of ACA to prevent tumor cell
metastatic invasion. The present study revealed that ACA impeded
glioblastoma cell migration by impairing the adhesive properties of
the cells, suggesting that ACA diminishes the invasive capacity and
growth of glioblastomas at secondary tumor sites in brain tissue by
blocking cell motility. Consistent with these data, In et
al(30) and Ichikawa et
al(17) also demonstrated that
ACA inhibited the migratory invasiveness of human oral carcinomas
in mouse xenografts and lung cancer cells, respectively.
Although the present study established that ACA is
an effective antagonist of glioblastoma cell proliferation and
migration, it is not likely to be a consequence of the ACA
antioxidant functions, as evidenced by ROS experimental data, which
revealed no change in the H2O2 concentration
between the control and glioblastoma cells treated with ACA.
Therefore the study also examined the mechanistic effects of ACA on
the cytokines, pro-inflammatory and tumorigenic activator molecules
of the JAK-STAT mitogenic pathway, which have been shown to be
upregulated by ROS (22) and act as
pro-survival molecules in brain tumors. Recent studies in
glioblastomas have demonstrated that the cytokine IL-6 plays a role
in enhancing glioblastoma cell survival and migratory invasion
(23–26), while studies in neuroblastomas
demonstrated that IL-6 and IL-1α promote cell survival by acting as
protectors of these nervous system-derived cancers (31). In contrast to the expected effects
of ACA on cytokine levels, in the present study, an increased
expression of IL-6 and IL-1α was observed in glioblastoma cells
treated with ACA. This provided evidence that indicated that
glioblastoma cells elicit a compensatory pro-survival response in
addition to a pro-apoptotic ACA-induced caspase 3 response.
To the best of our knowledge, this is the first
study that provides an insight into the function and versatility of
ACA to promote tumor cell death by circumventing the pro-survival
signaling mechanisms that are likely to contribute to the
therapeutic resistance of human cancers, in general and in
glioblastomas in particular. The novelty of ACA to overcome the
involvement of molecular signaling molecules associated with
perpetuating the maintenance and progression of human cancers make
it an attractive single and combinatorial drug agent for use in
continued experimental and clinical studies for the treatment of
this disease.
Acknowledgements
This study was supported by a grant
from the Louisiana Board of Regents (LEQSF 2012-13-ENH-UG-32). The
ACA was kindly provided by Dr Heather Kleiner of the Department of
Pharmacology, LSU Health Sciences Center (Shreveport, LA, USA).
References
|
1
|
Valko M, Rhodes CJ, Moncol J, Izakovic M
and Mazur M: Free radicals, metals and antioxidants in oxidative
stress-induced cancer. Chem Biol Interact. 160:1–40. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J and Yi J: Cancer cell killing via
ROS: to increase or decrease, that is the question. Cancer Biol
Ther. 7:1875–1884. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Acharya A, Das I, Chandhok D and Saha T:
Redox regulation in cancer: a double-edged sword with therapeutic
potential. Oxid Med Cell Longev. 3:23–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cabello CM, Bair WB III and Wondrak GT:
Experimental therapeutics: targeting the redox Achilles heel of
cancer. Curr Opin Investig Drugs. 8:1022–1037. 2007.PubMed/NCBI
|
|
5
|
Fang J, Seki T and Maeda H: Therapeutic
strategies by modulating oxygen stress in cancer and inflammation.
Adv Drug Deliv Rev. 61:290–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Montero AJ and Jassem J: Cellular redox
pathways as a therapeutic target in the treatment of cancer. Drugs.
71:1385–1396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pacher P, Nivorozhkin A and Szabó C:
Therapeutic effects of xanthine oxidase inhibitors: renaissance
half a century after the discovery of allopurinol. Pharmacol Rev.
58:87–114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kökoglu E, Belce A, Ozyurt E and Tepeler
Z: Xanthine oxidase levels in human brain tumors. Cancer Lett.
50:179–181. 1990.
|
|
9
|
Pu PY, Lan J, Shan SB, Huang EQ, Bai Y,
Guo Y and Jiang DH: Study of the antioxidant enzymes in human brain
tumors. J Neurooncol. 29:121–128. 1996.PubMed/NCBI
|
|
10
|
Ohnishi M, Tanaka T, Makita H, et al:
Chemopreventive effect of a xanthine oxidase inhibitor,
1′-acetoxychavicol acetate, on rat oral carcinogenesis. Jpn J
Cancer Res. 87:349–356. 1996.
|
|
11
|
Tanaka T, Kawabata K, Kakumoto M, et al:
Chemoprevention of azoxymethane-induced rat colon carcinogenesis by
a xanthine oxidase inhibitor, 1′-acetoxychavicol acetate. Jpn J
Cancer Res. 88:821–830. 1997.PubMed/NCBI
|
|
12
|
Ito K, Nakazato T, Murakami A, et al:
Induction of apoptosis in human myeloid leukemic cells by
1′-acetoxychavicol acetate through a mitochondrial- and
Fas-mediated dual mechanism. Clin Cancer Res. 10:2120–2130.
2004.
|
|
13
|
Ito K, Nakazato T, Xian MJ, et al:
1′-acetoxychavicol acetate is a novel nuclear factor kappaB
inhibitor with significant activity against multiple myeloma in
vitro and in vivo. Cancer Res. 65:4417–4424. 2005.
|
|
14
|
Campbell CT, Prince M, Landry GM, Kha V
and Kleiner HE: Pro-apoptotic effects of 1′-acetoxychavicol acetate
in human breast carcinoma cells. Toxicol Lett. 173:151–160.
2007.
|
|
15
|
Moffatt J, Hashimoto M, Kojima A, et al:
Apoptosis induced by 1′-acetoxychavicol acetate in Ehrlich ascites
tumor cells is associated with modulation of polyamine metabolism
and caspase-3 activation. Carcinogenesis. 21:2151–2157. 2000.
|
|
16
|
Ito K, Nakazato T, Murakami A, Ohigashi H,
Ikeda Y and Kizaki M: 1′-Acetoxychavicol acetate induces apoptosis
of myeloma cells via induction of TRAIL. Biochem Biophys Res
Commun. 338:1702–1710. 2005.
|
|
17
|
Ichikawa H, Takada Y, Murakami A and
Aggarwal BB: Identification of a novel blocker of I kappa B alpha
kinase that enhances cellular apoptosis and inhibits cellular
invasion through suppression of NF-kappa B-regulated gene products.
J Immunol. 174:7383–7392. 2005. View Article : Google Scholar
|
|
18
|
Muangnoi P, Lu M, Lee J, et al:
Cytotoxicity, apoptosis and DNA damage induced by Alpinia
galanga rhizome extract. Planta Med. 73:748–754. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Higashida M, Xu S, Kojima-Yuasa A, et al:
1′-Acetoxychavicol acetate-induced cytotoxicity is accompanied by a
rapid and drastic modulation of glutathione metabolism. Amino
Acids. 36:107–113. 2009.
|
|
20
|
Wu WS: The signaling mechanism of ROS in
tumor progression. Cancer Metastasis Rev. 25:695–705. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu WS, Wu JR and Hu CT: Signal cross talks
for sustained MAPK activation and cell migration: the potential
role of reactive oxygen species. Cancer Metastasis Rev. 27:303–314.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Naik E and Dixit VM: Mitochondrial
reactive oxygen species drive proinflammatory cytokine production.
J Exp Med. 208:417–420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saidi A, Hagedorn M, Allain N, et al:
Combined targeting of interleukin-6 and vascular endothelial growth
factor potently inhibits glioma growth and invasiveness. Int J
Cancer. 125:1054–1064. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li R, Li G, Deng L, Liu Q, Dai J, Shen J
and Zhang J: IL-6 augments the invasiveness of U87MG human
glioblastoma multiforme cells via up-regulation of MMP-2 and
fascin-1. Oncol Rep. 23:1553–1559. 2010.PubMed/NCBI
|
|
25
|
Liu Q, Li G, Li R, et al: IL-6 promotion
of glioblastoma cell invasion and angiogenesis in U251 and T98G
cell lines. J Neurooncol. 100:165–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Michaud-Levesque J, Bousquet-Gagnon N and
Béliveau R: Quercetin abrogates IL-6/STAT3 signaling and inhibits
glioblastoma cell line growth and migration. Exp Cell Res.
318:925–935. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanriverdi T, Hanimoglu H, Kacira T, et
al: Glutathione peroxidase, glutathione reductase and protein
oxidation in patients with glioblastoma multiforme and transitional
meningioma. J Cancer Res Clin Oncol. 133:627–633. 2007. View Article : Google Scholar
|
|
28
|
Dokic I, Hartmann C, Herold-Mende C and
Régnier-Vigouroux A: Glutathione peroxidase 1 activity dictates the
sensitivity of glioblastoma cells to oxidative stress. Glia.
60:1785–1800. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Atukeren P, Kemerdere R, Kacira T, et al:
Expressions of some vital molecules: glioblastoma multiforme versus
normal tissues. Neurol Res. 32:492–501. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
In LL, Arshad NM, Ibrahim H, Azmi MN,
Awang K and Nagoor NH: 1′-Acetoxychavicol acetate inhibits growth
of human oral carcinoma xenograft in mice and potentiates cisplatin
effect via proinflammatory microenvironment alterations. BMC
Complement Altern Med. 12:1792012.
|
|
31
|
Bissonnette CJ, Klegeris A, McGeer PL and
McGeer EG: Interleukin 1alpha and interleukin 6 protect human
neuronal SH-SY5Y cells from oxidative damage. Neurosci Lett.
361:40–43. 2004. View Article : Google Scholar : PubMed/NCBI
|