Introduction
The Ewing’s sarcoma family of tumors (ESFT) are
highly malignant, metastatic, primitive small round cell tumors of
the bone and soft tissue that affect children and adolescents. ESFT
comprises morphologically heterogeneous tumors that are
characterized by non-random chromosomal translocations between the
EWS gene on chromosome 22q12 and one of several members of the
E-twenty six (ETS) family of transcription factors. In ∼85% of
cases of ESFT, the FLI1 gene on chromosome 11 is the fusion partner
of EWS (EWSFLI1) (1,2); in ∼10%, the EWS fusion partner is the
ERG gene on chromosome 22 (EWS-ERG) (3–5). Other
ESFT family members have been identified as fusion partners of EWS,
but these cases are rare. In <1% of cases, variant
translocations, namely t(7;22)(p22;q12), t(17;22)(q12;q12) and
t(2;22) (q33;q12), involving fusion of the EWS gene with ETV1, E1AF
(also known as ETV4) and FEV genes, respectively, have been
described (6–9).
Dramatic improvements in the survival of ESFT have
been achieved for children and adolescents due to the development
of multidisciplinary treatments, including multiple drug
chemotherapy, refined surgical techniques and appropriate radiation
therapy. Between 1975 and 2002, the 5-year survival rate has
increased from 59 to 76% for children younger than 15 years and
from 20 to 49% for adolescents aged 15 to 19 years (10). However, the current studies show
that 30–40% of non-metastatic ESFT patients will still develop
recurrent disease (local and/or metastatic) in spite of
multidisciplinary treatment (11).
The known prognostic factors for ESFT are tumor size or volume,
serum lactate dehydrogenase (LDH) levels, axial localization and
age (>15 years). Under treatment, a poor histological response
to preoperative chemotherapy and incomplete or no surgery for local
therapy are further adverse prognostic factors (12).
Here, a case of ESFT of the femur with an atypical
clinical course is reported. At the local hospital, the patient
received inadequate initial treatment which consisted of tumor
curettage, chemotherapy with insufficient dose intensity and
low-dose radiation therapy. In spite of the inadequate initial
treatment, the patient had been disease-free for the subsequent 18
years. The patient was examined due to thigh pain following local
recurrence 18 years after initial treatment and received standard
multimodal therapy employing combination chemotherapy and wide
surgical excision. After receiving treatment, the patient showed no
evidence of disease for the next 9 years. The study was approved by
the Ethics Committee of Mie University, Tsu City, Japan. Written
informed consent was obtained from the patient.
Case report
A 38-year-old male presented with pain in his right
thigh. The patient had a past history of treatment for a bone tumor
of the right proximal femur. At 20 years of age, the patient had
experienced right thigh pain and consulted a doctor at the local
hospital. The patient was noted to have a bone tumor of the right
femoral bone without distant metastasis and underwent surgical
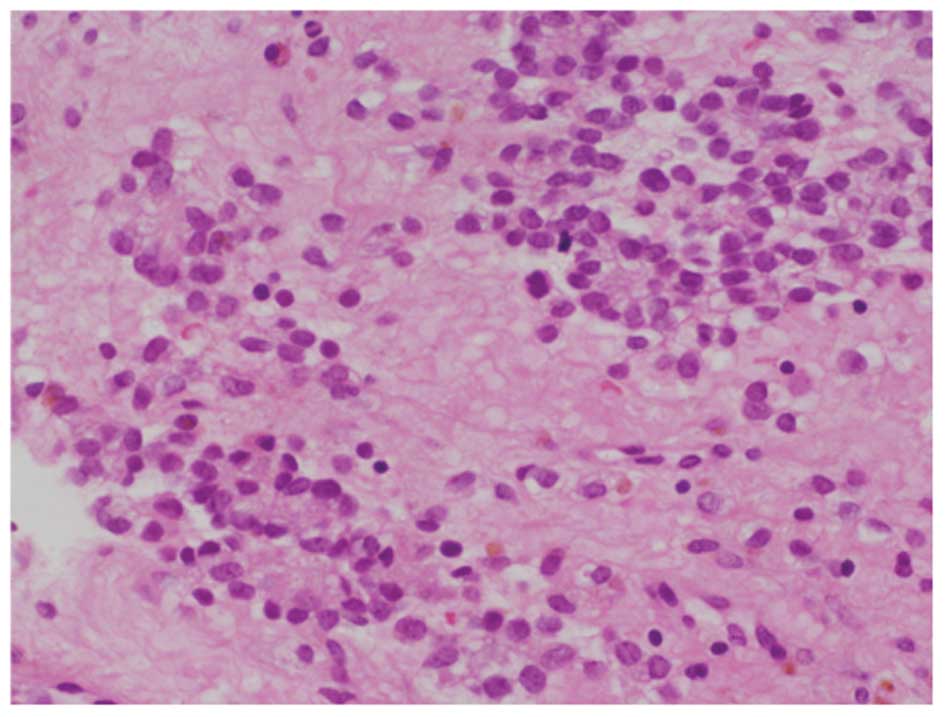
curettage of the tumor. The specimen revealed a malignant small
round cell neoplasm with regular nuclear contours, finely dispersed
chromatin and scanty cytoplasm without prominent nucleoli (Fig. 1). The patient received one cycle of
postoperative chemotherapy that consisted of vincristine and
cyclophosphamide and local irradiation with a total dose of 40 Gy.
Two years after the initial operation (at 22 years of age), the
patient developed severe pain in his right thigh after braking hard
while driving. The radiographs taken at the time showed a fracture
of the femur where the bone tumor had been located. Open reduction
and internal fixation (ORIF) using intramedullary nails was
performed. One year after the ORIF, the patient stubbed his toe and
could not walk due to severe right thigh pain. Radiographs showed
an undisplaced femoral re-fracture. He was conservatively treated
and subsequent roentgenograms demonstrated bone union. His
subsequent postoperative course had been uneventful for 15
years.
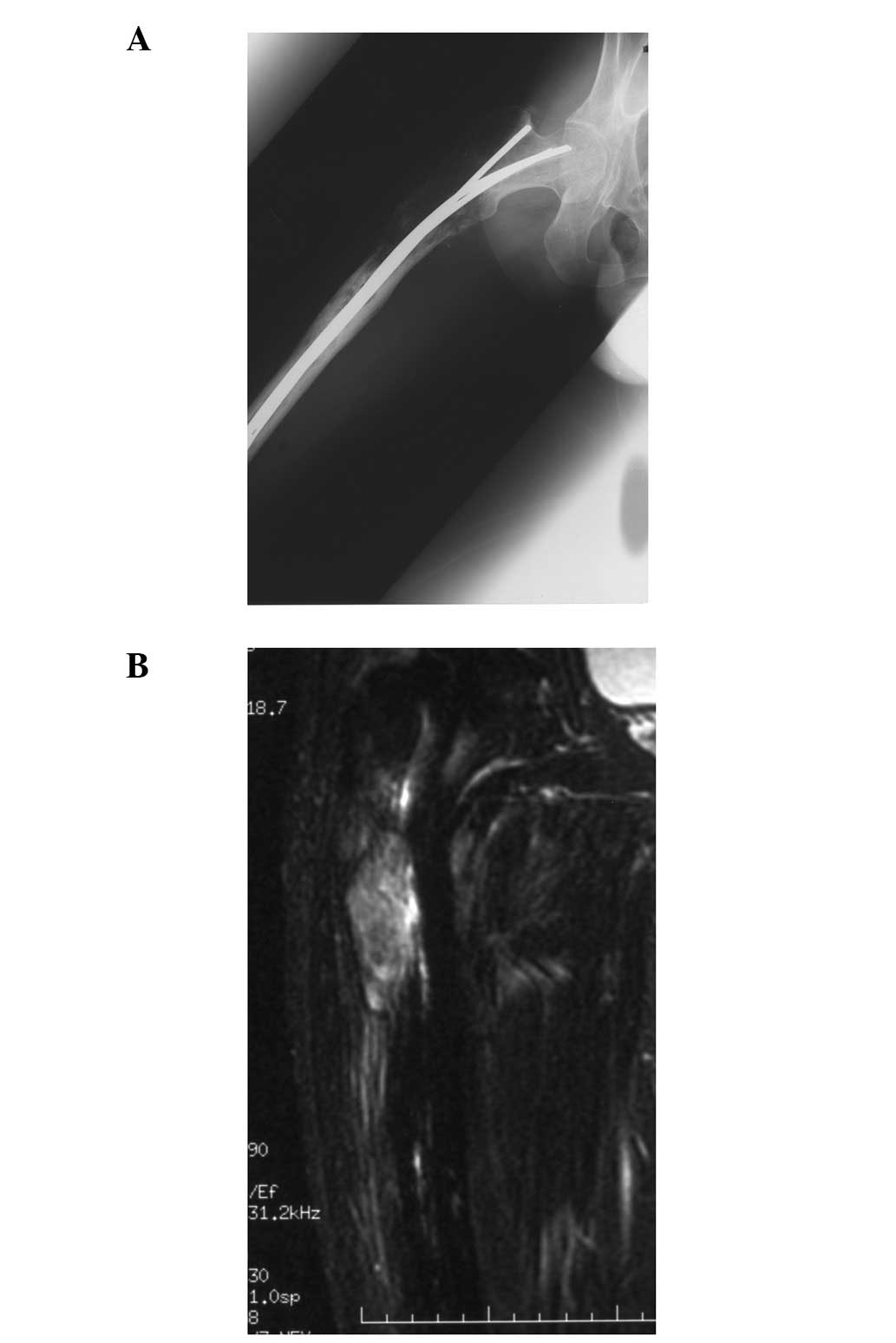
At 38 years of age, the patient was referred with
right thigh pain that had persisted for several months. Radiographs
showed a lytic lesion from the trochanteric area to the proximal
diaphysis (Fig. 2A). Magnetic
resonance images showed an intramedullary lesion with an
extraskeletal mass (Fig. 2B) and
further metastatic work-up was negative. As local recurrence was
suspected, an open biopsy from the intertrochanteric lesion was
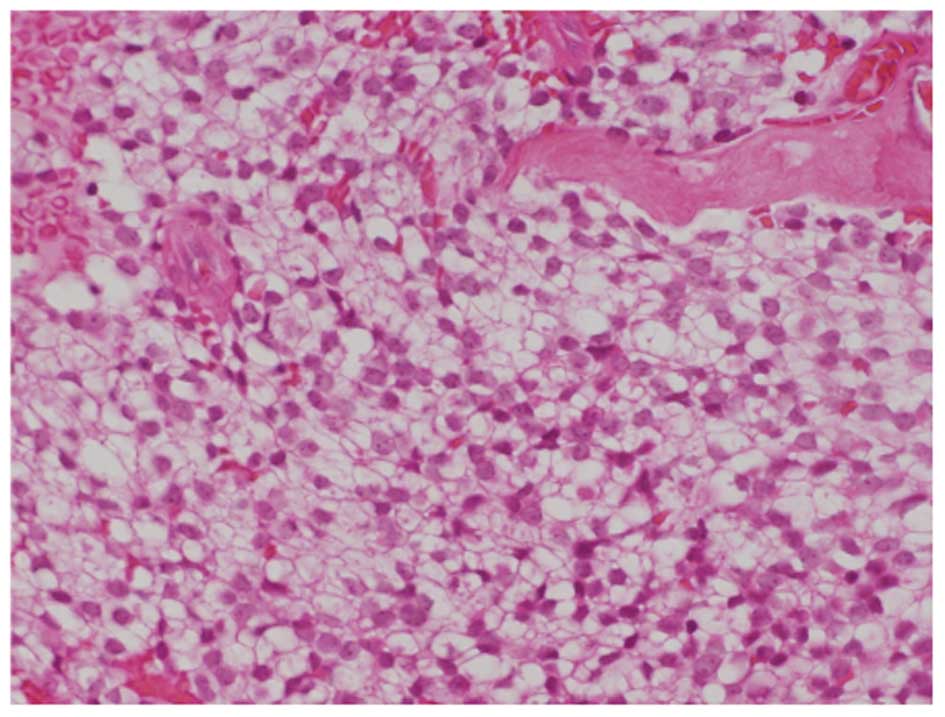
performed. Microscopic findings revealed a malignant small round
cell neoplasm with regular nuclear contours and finely dispersed
chromatin without prominent nucleoli (Fig. 3). The microscopic findings were
similar to those obtained at first surgery, performed at 20 years
of age. Immunohistochemical and histochemical staining showed that
the neoplasm was positive for glycogen (PAS), neuron-specific
enolase (NSE), MIC-2 and S-100 protein, and negative for α-SMA,
CD34, CD56, chromogranin A, neurofilament and vimentin. The MIB-1
index was <10%. Although molecular biological investigations did
not reveal the presence of any characteristic fusion genes,
including EWS-FLI1, EWS-ERG, EWS-FEV, EWS-ETV1 and EWS-E1AF, a
final diagnosis of a local recurrence of ESFT was made. Based on
the diagnosis of ESFT, the patient underwent chemotherapy
consisting of vincristine, doxorubicin, cyclophosphamide and
ifosfamide (12) as well as wide
resection of the tumor, combined with reconstruction using a
prosthesis. The microscopic findings of the specimen from the
widely resected proximal femur revealed a minimum response to the
preoperative chemotherapy (necrotic rate <10%). The patient has
been disease-free for the past 9 years following this surgery.
Discussion
ESFT is the second most common primary malignant
bone tumor in children and adolescents. Recently, the treatment
outcome of ESFT has been improved significantly with the use of
multimodal therapy. However, 30–40% of patients with localized
disease and 80% of those with metastatic disease succumb due to
disease progression.
The present case is unusual for a number of reasons.
Firstly, the patient received inadequate initial treatment at the
local hospital which consisted of tumor curettage, chemotherapy
with an insufficient dose intensity and low-dose radiation therapy.
In spite of such inadequate initial treatment, the patient had been
disease-free for the subsequent 18 years. The known prognostic
factors for ESFT are tumor size or volume, serum LDH levels, axial
localization and age (>15 years). Under treatment, a poor
histological response to preoperative chemotherapy and incomplete
or no surgery for local therapy are adverse prognostic factors
(12). One possible reason for the
patient being disease-free for the 18 years following the initial
treatment is that the radiation therapy may have been extremely
effective for this ESFT.
Secondly, the local recurrence occurred 18 years
after initial treatment. Local recurrence of malignant bone tumors
commonly occurs within the first 1–2 years (14,15)
and usually occurs within 3 years of the initial treatment
(16,17). Since ESFT is a highly malignant
aggressive tumor, local recurrence over fifteen years later is
extremely rare. Bacci et al (18) reported that 187 of 215 patients
(87.0%) relapsed within the first 5 years after starting treatment,
while 28 (13.0%) relapsed after more than 5 years. In an analysis
of 402 patients followed up for a median of 17.7 years, the longest
time from definitive surgery to local recurrence was 7.0 years
(18). Donati et al
(19) reported the longest time
until recurrence to be 3.3 years in an analysis of 56 cases.
Gasparini et al (20)
described the long-term outcome in 121 patients with monostotic
ESFT treated with combined modality therapy. The mean follow-up
time in their study was 12 years, with one patient developing local
recurrence at 14 years. No other cases with local recurrence of
more than 15 years after the initial treatment were found, except
for one case who relapsed 17 years after diagnosis (21).
Thirdly, the microscopic findings of the specimen
from the widely resected proximal femur at the most recent surgery
revealed only a minimum response to the preoperative chemo-therapy.
Despite this, the patient has been disease-free for the past 9
years. Lin et al reported that the histological response to
preoperative chemotherapy is a strong predictor of local
recurrence. Patients who have a poor response (<90% necrosis)
have a 50% risk of local recurrence at 5 years (22). Patients with bone or bone marrow
metastases and patients with recurrent disease still fare poorly,
with 5-year survival rates of 20% (10). This result suggests that the ESFT of
our patient had nonmetastatic and non-aggressive biological
behavior.
Finally, although the microscopic,
immunohistochemical and histochemical findings were typical of
ESFT, the molecular biological investigation did not reveal any of
the characteristic fusion genes, including EWS-FLI1, EWS-ERG,
EWS-FEV, EWS-ETV1 or EWS-E1AF. The diagnosis of the tumor was made
using several modalities, such as light microscopy and
immunohistochemistry, and was confirmed by several pathologists
with specialization in musculoskeletal tumors. The current case,
with its unusual clinical course, may involve an unknown fusion
gene.
In consideration of both the good clinical course
unusual for ESFT and the absence of any of the previously reported
fusion genes, the present case may be a rare subtype of ESFT with
an unknown chromosomal translocation and relatively non-aggressive
biological behavior. Further genetic investigation is therefore
required for this patient.
References
|
1.
|
Delattre O, Zucman J, Plougastel B, et al:
Gene fusion with an ETS DNA-binding domain caused by chromosome
translocation in human tumours. Nature. 359:162–165. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Arvand A and Denny CT: Biology of EWS/ETS
fusions in Ewing’s family tumors. Oncogene. 20:5747–5754. 2001.
|
|
3.
|
Delattre O, Zucman J, Melot T, et al: The
Ewing family of tumors - a subgroup of small-round-cell tumors
defined by specific chimeric transcripts. N Engl J Med.
331:294–299. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Sorensen PH, Lessnick SL, Lopez-Terrada D,
Liu XF, Triche TJ and Denny CT: A second Ewing’s sarcoma
translocation, t(21;22), fuses the EWS gene to another ETS-family
transcription factor, ERG. Nat Genet. 6:146–151. 1994.
|
|
5.
|
Zucman J, Melot T, Desmaze C, et al:
Combinatorial generation of variable fusion proteins in the Ewing
family of tumours. EMBO J. 12:4481–4487. 1993.PubMed/NCBI
|
|
6.
|
Peter M, Couturier J, Pacquement H, et al:
A new member of the ETS family fused to EWS in Ewing tumors.
Oncogene. 14:1159–1164. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kaneko Y, Yoshida K, Handa M, et al:
Fusion of an ETS-family gene, E1AF, to EWS by t(17;22)(q12;q12)
chromosome trans-location in an undifferentiated sarcoma of
infancy. Genes Chromosomes Cancer. 15:115–121. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Urano F, Umezawa A, Hong W, Kikuchi H and
Hata J: A novel chimera gene between EWS and E1A-F, encoding the
adenovirus E1A enhancer- binding protein, in extraosseous Ewing’s
sarcoma. Biochem Biophys Res Commun. 219:608–612. 1996.PubMed/NCBI
|
|
9.
|
Jeon IS, Davis JN, Braun BS, et al: A
variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to
the ETS gene ETV1. Oncogene. 10:1229–1234. 1995.
|
|
10.
|
Smith MA, Seibel NL, Altekruse SF, et al:
Outcomes for children and adolescents with cancer: challenges for
the twenty-first century. J Clin Oncol. 28:2625–2634. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Bacci G, Ferrari S, Longhi A, et al:
Therapy and survival after recurrence of Ewing’s tumours: the
Rizzoli experience in 195 patients treated with ajuvant and
neoajuvant chemotherapy from 1979 to 1997. Ann Oncol. 14:1654–1659.
2003.
|
|
12.
|
Paulussen M, Bielack S, Jürgens H and
Casali PG; ESMO Guidelines Working Group: Ewing’s sarcoma of the
bone: ESMO clinical recommendations for diagnosis,treatment and
follow-up. Ann Oncol. 20:140–142. 2009.
|
|
13.
|
Rosito P, Mancini AF, Rondelli R, et al:
Italian Cooperative Study for the treatment of children and young
adults with localized Ewing sarcoma of bone: a preliminary report
of 6 years of experience. Cancer. 86:421–428. 1999.PubMed/NCBI
|
|
14.
|
McTiernan AM, Cassoni AM, Driver D, et al:
Improving Outcomes After Relapse in Ewing’s Sarcoma: Analysis of
114 Patients From a Single Institution. Sarcoma.
2006:835482006.PubMed/NCBI
|
|
15.
|
Grier HE, Krailo MD, Tarbell NJ, et al:
Addition of ifosfamide and etoposide to standard chemotherapy for
Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl
J Med. 348:694–701. 2003.
|
|
16.
|
Indelicato DJ, Keole SR, Shahlaee AH, et
al: Long-term clinical and functional outcomes after treatment for
localized Ewing’s tumor of the lower extremity. Int J Radiat Oncol
Biol Phys. 70:501–509. 2008.PubMed/NCBI
|
|
17.
|
Rodríguez-Galindo C, Liu T, Krasin MJ, Wu
J, et al: Analysis of prognostic factors in ewing sarcoma family of
tumors: review of St. Jude Children’s Research Hospital studies.
Cancer. 110:375–384. 2007.
|
|
18.
|
Bacci G, Forni C, Longhi A, et al:
Long-term outcome for patients with non-metastatic Ewing’s sarcoma
treated with adjuvant and neoadjuvant chemotherapies. 402 patients
treated at Rizzoli between 1972 and 1992. Eur J Cancer. 40:73–83.
2004.
|
|
19.
|
Donati D, Yin J, Di Bella C, et al: Local
and distant control in non-metastatic pelvic Ewing’s sarcoma
patients. J Surg Oncol. 96:19–25. 2007.PubMed/NCBI
|
|
20.
|
Gasparini M, Lombardi F, Ballerini E, et
al: Long-term outcome of patients with monostotic Ewing’s sarcoma
treated with combined modality. Med Pediatr Oncol. 23:406–412.
1994.PubMed/NCBI
|
|
21.
|
McLean TW, Hertel C, Young ML, et al: Late
events in pediatric patients with Ewing sarcoma/primitive
neuroectodermal tumor of bone: the Dana-Farber Cancer
Institute/Children’s Hospital experience. J Pediatr Hematol Oncol.
2:486–493. 1999.PubMed/NCBI
|
|
22.
|
Lin PP, Jaffe N, Herzog CE, et al:
Chemotherapy response is an important predictor of local recurrence
in Ewing sarcoma. Cancer. 109:603–611. 2007. View Article : Google Scholar : PubMed/NCBI
|