Introduction
The sirtuins, or SIRTs, are highly conserved
mammalian homologues of yeast silent mating-type information
regulation 2, homolog (Sir2), which catalyze
NAD+-dependent histone deacetylation and ADP
ribosylation (1). Numerous studies
have shown that the levels of silent mating-type information
regulation 2, homolog 1 (SIRT1) are significantly elevated in
prostate, ovarian, gastric and colorectal cancer, as well as
hepatocellular carcinoma (2–6).
Moreover, SIRT1 inhibition has been reported to suppress cell
growth and induce cell cycle arrest or apoptosis in cancer cells
(7). Although SIRT1 has emerged as
a key regulator in various cellular pathways, the regulatory
mechanisms responsible for SIRT1 activity have not been determined.
SIRT1, the mammalian homolog of Sir2, has been shown to regulate a
wide variety of cellular processes (8,9),
including glucose metabolism (10,11),
the cell cycle, growth and differentiation, inflammation,
senescence, apoptosis (12), the
stress response (13) and
aging.
The present study focused on the role of SIRT1 in
the stress response. Previous studies have shown that Sir2
represses p53-dependent apoptosis in response to DNA damage and
oxidative stress by physically interacting with p53 (13) and the forkhead transcription factor
(FOXO) family of proteins (14),
indicating that SIRT1 promotes cellular survival. However,
embryonic stem cells and fibroblasts from SIRT1-null mice showed no
altered resistance to DNA damage-induced stress (15). In tumor cells, SIRT1 also failed to
alter cell survival following DNA damage (16).
The controversial functions of SIRT1 require
investigation as to i) whether SIRT1 has biological function in
tumor cells subjected to antitumor agent treatment; ii) how SIRT1
executes its function during the stress response; and iii) what
would happen to tumor cells if the deacetylase activity of SIRT1
was inhibited. For the present study, nicotinamide (NAM), the most
potent inhibitor of Sir2 enzymes to date (17–20),
was used to inhibit the deacetylase activity of SIRT1.
Materials and methods
Cell culture and reagents
Human breast cancer MCF-7 cells were seeded at
1×105 cells/well (n=2 for each condition) in 24-well
tissue-culture plates containing 0.5 ml complete medium (RPMI-1640
medium supplemented with 10% fetal bovine serum; Tianjin Haoyang
Biotech Company, Tianjin, China) and 2 mM glutamine, penicillin and
streptomycin (100 units/ml). The cells were incubated at 37°C in a
humidified atmosphere with 5% CO2. The study was
approved by the ethics committee of China Medical University
(Shenyang, P.R. China).
NAM was prepared as a 1 M solution with
phosphate-buffered saline (PBS) and stored at −20°C ready for use.
Doxorubicin (doxo), arsenic trioxide (As2O3)
and Taxol were purchased from Sigma-Aldrich Chemical Inc. (St.
Louis, MO, USA).
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazo lium bromide
(MTT), Hoechst 33342, RNase A, RPMI-1640 medium and DMEM were
purchased from Invitrogen (Carlsbad, CA, USA).
Cell viability assay
The MTT assay was used to assess cell viability. The
cells were seeded into 96-well plates (4,000 cells/well) for 24 h
and treated with various anti-tumor agents for 72 h. Subsequently,
20 μl MTT (5 mg/ml) was added to the medium. Subsequent to
incubation at 37°C for 4 h, the culture medium containing MTT was
removed and 200 μl DMSO was added to solubilize the blue
formazan formed by the viable cells. The plates were read with an
ELISA plate reader at 570 nm. Cell viability is presented as a
percentage ratio of exposed cells to control cells.
Immunoblot analysis
The treated cells were scraped from the culture,
washed with PBS and lysed with buffer containing 25 mM Tris-HCl (pH
7.5) 150 mM NaCl, 2 mM EDTA, 10% glycerol, 10 mM glycerophosphate,
5 mM sodium pyrophosphate, 5 mM NaF, 1 mM
Na3VO4, 0.5% Triton X-100, 1 mM PMSF, 2
μg/ml aprotinin and 2 μg/ml leupeptin. Equal amounts
of protein samples were loaded onto SDS-PAGE gels and transferred
to PVDF membranes, then probed with the corresponding antibodies.
Antibodies directed against poly(ADP-ribose) polymerase (PARP),
cleaved caspase 6, cleaved caspase 7 and cleaved caspase 9 were
obtained from Cell Signaling Technology (Beverly, MA, USA), while
anti-SIRT1 antibody was purchased from Santa Cruz Biotechnology
Inc. (Santa Cruz, CA, USA). The protein signals were detected with
an enhanced chemiluminescence system (RPN 2106) according to the
manufacturer’s instructions (Amersham Biosciences, Indianapolis,
IN, USA).
Detection of chromatin condensation with
Hoechst 33342 staining
The cells (2×105 per well) were seeded
into a six-well plate for 24 h and treated with NAM for the
indicated times. The suspended and adherent cells were collected
and incubated with 2 μg/ml Hoechst 33342 at 37°C for 30 min.
Chromatin condensation was observed by fluorescence microscopy.
Cell cycle analysis
The treated cells were trypsinized and washed once
with PBS, then fixed with cold 75% ethanol overnight. The fixed
cells were washed twice with PBS and incubated with 100
μg/ml RNase I and 50 mg/ml propidium iodide (PI) for 30 min,
then the cellular DNA content was determined by flow cytometry
(FACS Calibur, BD Biosciences, Franklin Lakes, NJ, USA).
Assessment of apoptosis by Annexin
V+/PI− staining
The apoptotic cells were detected by the Apop
NexinTM FITC Apoptosis Detection kit (Chemicon,
Temecula, CA, USA) according to the manufacturer’s instructions.
Briefly, the suspended and adherent cells were pooled, washed twice
with ice-cold PBS and resuspended in binding buffer to
106/ml. Next, 0.2 ml of this cell suspension was
incubated with 3 μl fluorescein isothiocyanate
(FITC)-labeled Annexin V and 2 μl PI for 60 min at room
temperature in the dark. Samples were analyzed by flow
cytometry.
Statistical analysis
Each experiment was repeated at least three times.
Data are presented as the mean ± standard deviation.
The combined effects of the antitumor agents and NAM
on cell viability were calculated according to the coefficient of
drug interaction (CDI) (21),
calculated by the formula: CDI = AB / (A × B), where AB represents
the cell viability of the combination of drug A and B, while A or B
represent cell viability of the single compound alone. CDI <1,
CDI = 1 and CDI >1 represent the synergy, additivity and
antagonism of A and B, respectively.
Results
SIRT1 expression increases under certain
stress response levels
In order to study the biological functions of SIRT1
in the stress response caused by antitumor agent treatment,
As2O3, Taxol and doxo, the most commonly used
clinical antitumor agents, were selected according to their
different mechanisms of action. As2O3 is an
effective therapy in acute promyelocytic leukemia (APL) (22) and has also exhibited promising
activities in other hematological and solid tumors (23,24).
As2O3 targets differentiation, apoptosis and
protein oxidative damage (25).
Taxol binds microtubules and causes the kinetic suppression of
microtubule dynamics, thus killing cancer cells through the
induction of apoptosis (26,27).
Topoisomerase II is generally recognized to be the main cellular
target of doxo. There appears to be general agreement that
oxidative stress is a significant contributor to the antitumor
activity of doxo (28,29).
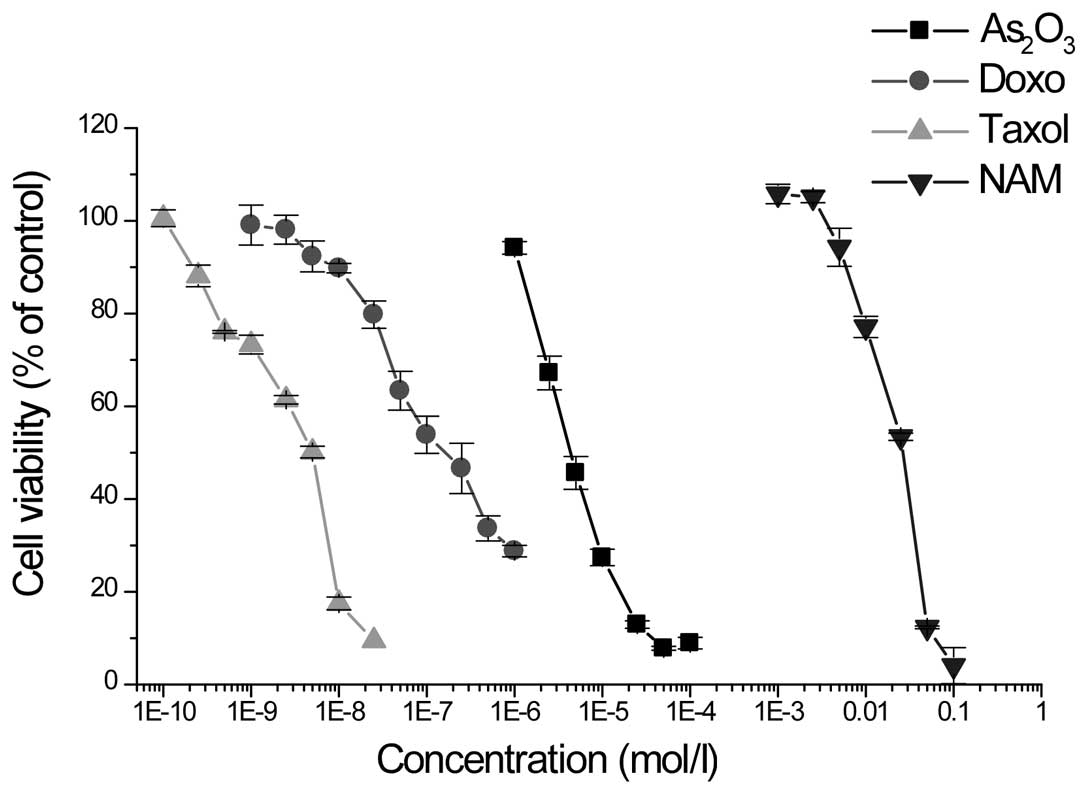
First, the anti-proliferative effects of doxo,
Taxol, As2O3 and NAM were examined with the
MTT assay. Incubation of MCF-7 cells with various concentrations of
doxo, Taxol, As2O3 and NAM led to the
dose-dependent inhibition of cell proliferation (Fig. 1). The IC50 values
obtained subsequent to 72 h of treatment were
(1.7±0.1)×10−7, (2.9±0.2)×10−9,
(5.4±0.4)×10−6 and (21.5±0.7)×10−3 mol/l for
doxo, Taxol, As2O3 and NAM, respectively.
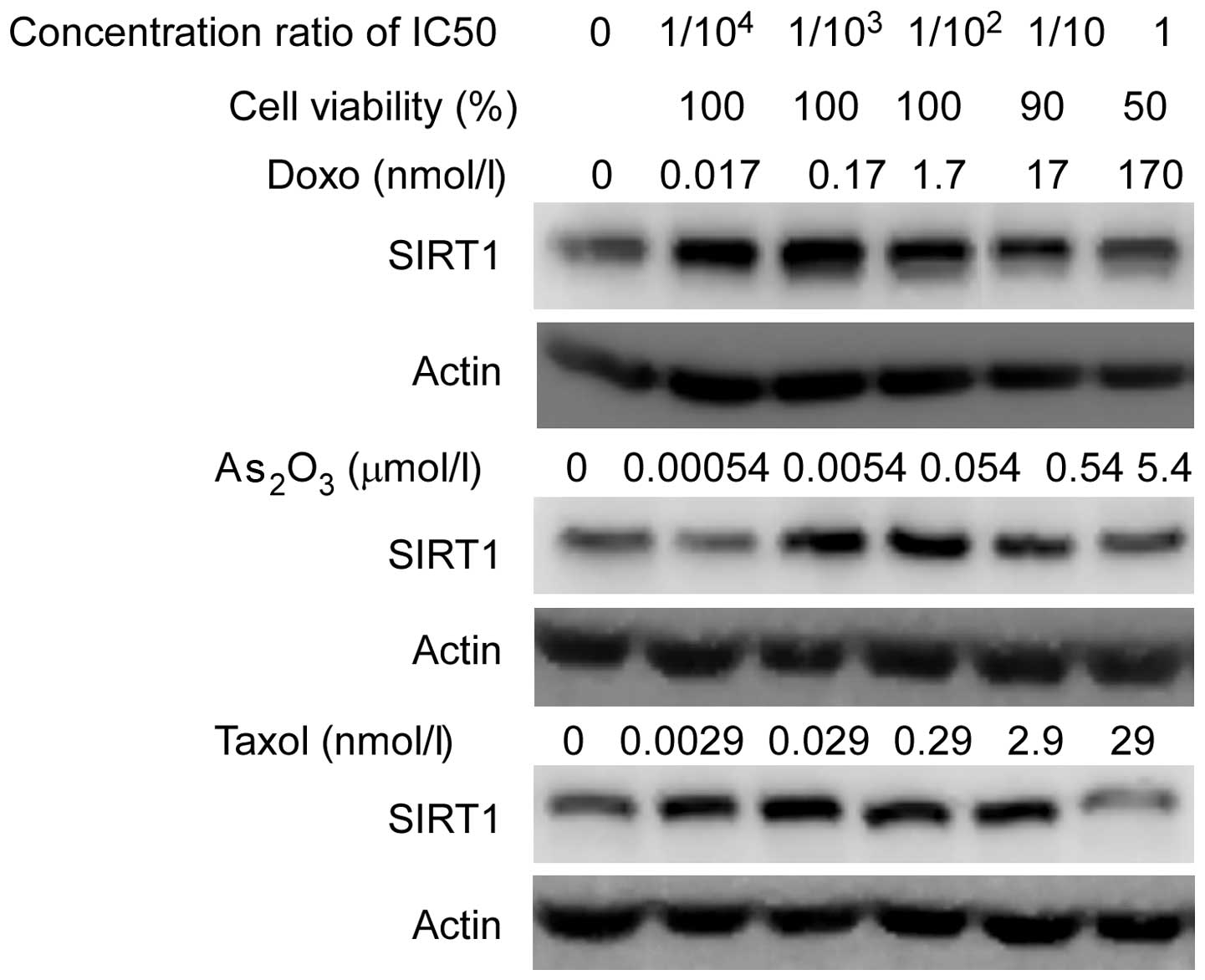
Subsequently, the MCF-7 cells were treated with
doxo, Taxol and As2O3 at various
concentrations, which led to 100%, 90% and 50% cell viability,
respectively, and SIRT1 expression was detected by immunoblot
analysis following antitumor agent treatment for 24 h. Notably,
increased expression levels of SIRT1 were observed at low
concentrations (>90% cell viability), but not at high drug
concentrations (50% cell viability; Fig. 2). This result may suggest that only
the stress levels that lightly damage tumor cells are able to
activate the SIRT1 pathway.
NAM decreases the viability of MCF-7
cells through SIRT1 deacetylases
It is well known that SIRT1 is a nuclear enzyme with
deacetylase activity, and the present study aimed to investigate
what happened if SIRT1 deacetylates were inhibited during the
stress response. Therefore, NAM was added during antitumor agent
treatment to inhibit SIRT1 deacetylase, and the change in cell
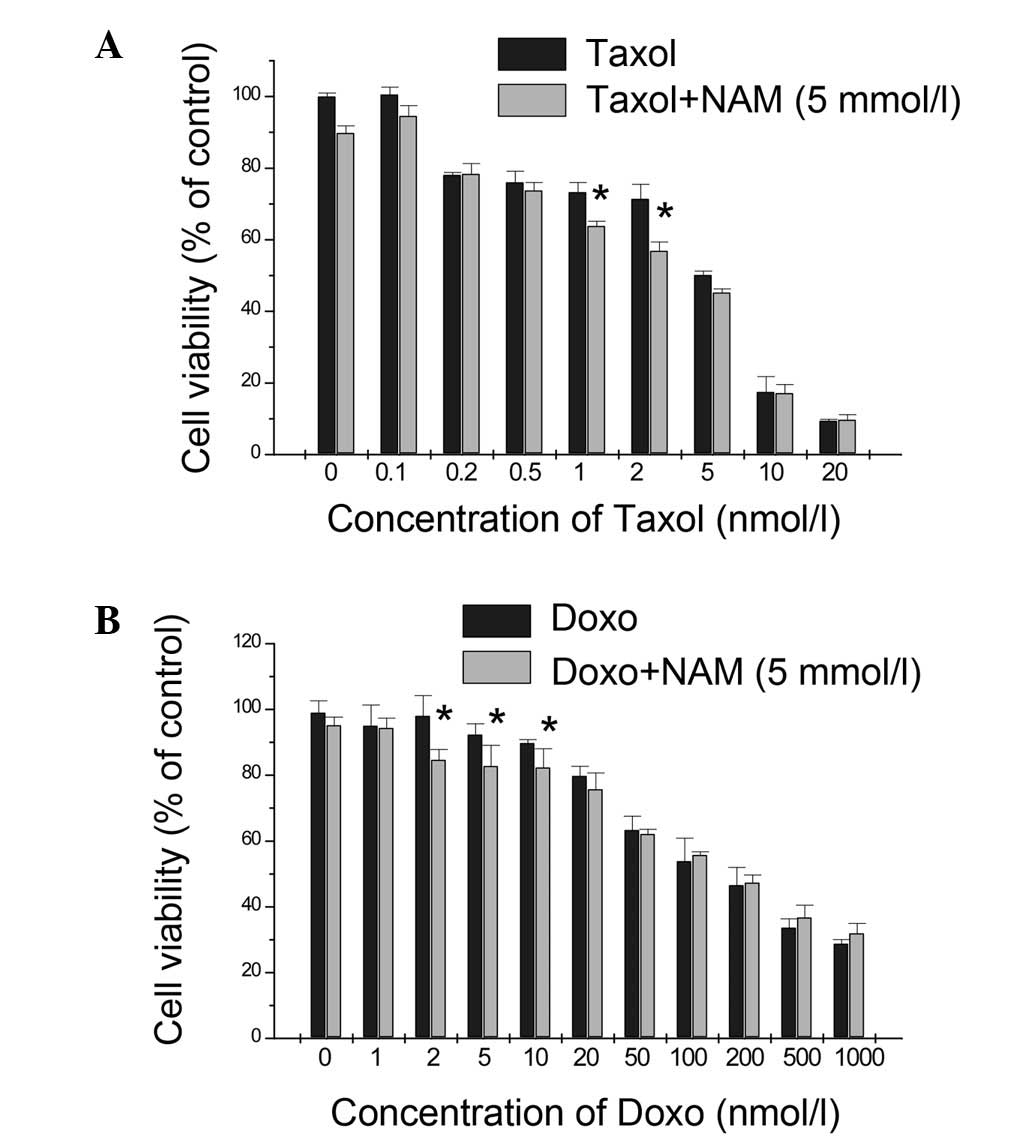
viability was detected with the MTT assay. NAM at 5 mM was selected
to inhibit SIRT1 deacetylase since it is non-toxic and remains
active at this concentration (17–20).
It is worth noting that NAM decreased the viability of the MCF-7
cells exposed to doxo and Taxol at low drug concentrations and that
NAM had a synergistic effect only with 1 and 2.5 nmol/l Taxol (CDI,
0.96 and 0.88, respectively; Fig.
3A), while a similar result was obtained with the combined
usage of doxo and NAM (CDI<1; Fig.
3B). This result indicates that SIRT1 promotes cancer cell
survival under certain conditions of applied cellular stress and
that this activity is mediated by its deacetylase activity.
Apoptosis is induced by NAM in MCF-7
cells
The subsequent aim was to investigate whether the
inhibition of the deacetylase activity of SIRT1 with NAM was able
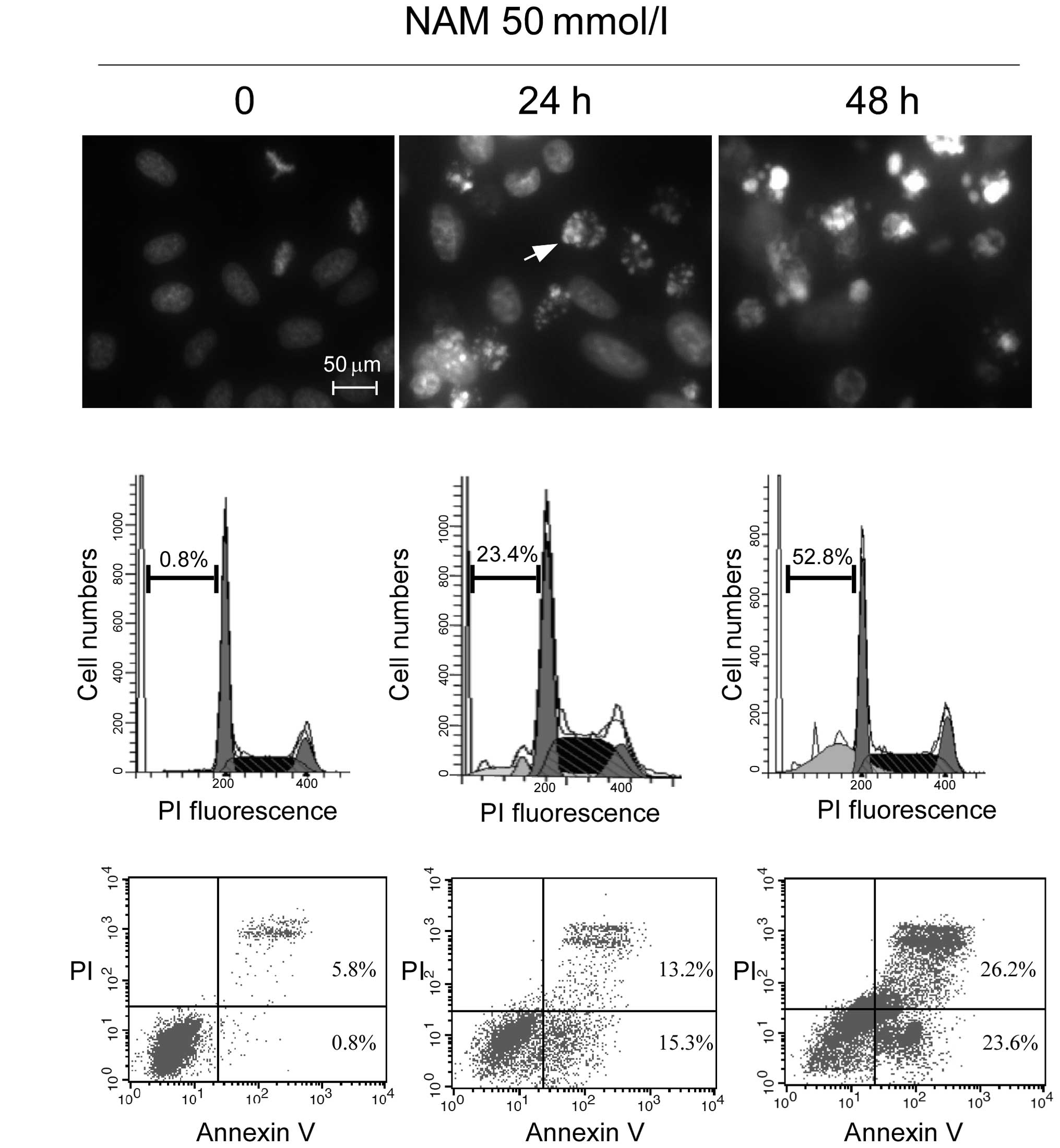
to induce apoptosis. MCF-7 cells were exposed to 50 mmol/l NAM for
24 and 48 h, and typical biochemical hallmarks of apoptosis
(30), such as chromatin
condensation (Fig. 4A), sub
G1 cell cycle distribution and Annexin
V+/PI− stained cells, were detected to
demonstrate the occurrence of apoptosis. In Fig. 4A, a considerable amount of chromatin
condensation and apoptotic bodies (as indicated by the white arrow)
were observed in the NAM-treated cells assessed by fluorescence
microscopy. For the flow cytometry, an increased sub-G1
cell cycle population (Fig. 4B) and
a greater number of Annexin V+/PI− staining
cells (Fig. 4C) were detected in
the NAM-treated cells. These data indicated that NAM induces
typical apoptotic features in MCF-7 cells.
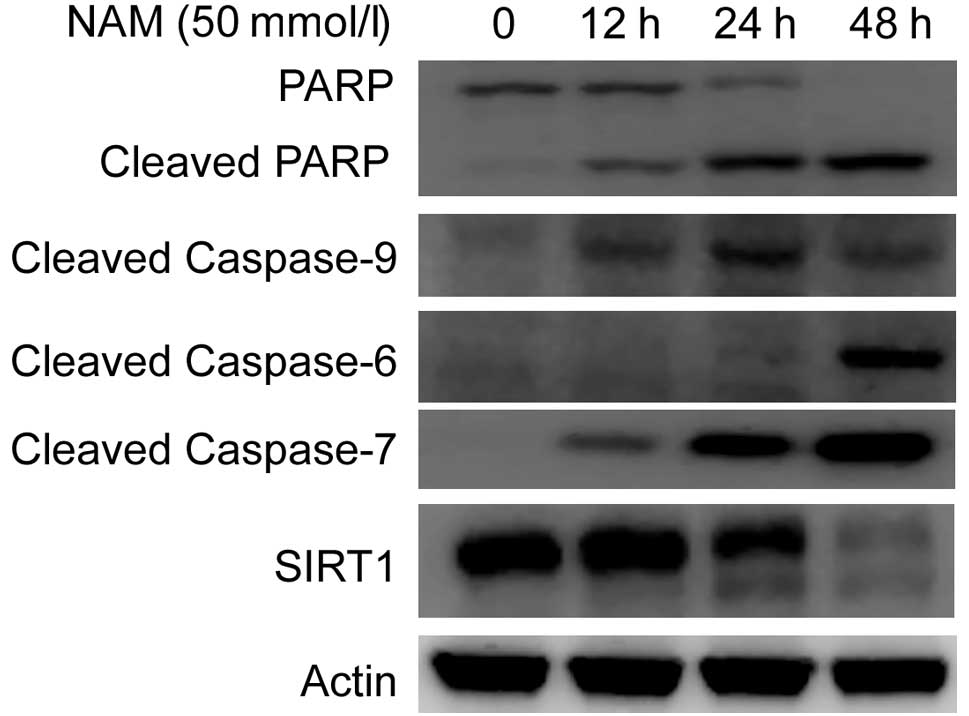
Apoptosis is a tightly controlled multi-step process
of cell death, with the orderly involvement of proteins, such as
initiator caspase 9 and executioner caspases 6 and 7, which then
cleave PARP when activated. To determine the role of the caspase
cascade in NAM-induced apoptosis, the MCF-7 cells were exposed to
50 mmol/l NAM for 12, 24 and 48 h, respectively. The activation of
caspases and PARP was detected by cleavage fragments in the
immunoblot analysis. The results (Fig.
5) showed that NAM triggered the activation of caspases 9, 6
and 7 and the cleavage of PARP in a time-dependent manner.
Together, this led to the conclusion that NAM induces apoptosis in
tumor cells, accompanied by the activation of the caspase
cascade.
Discussion
In the present study, SIRT1 showed an increased
expression with low concentrations of drug treatment, but no
altered expression at high concentrations. We propose that
different drug concentrations may cause different degrees of
cellular damage, which arouse various biological effects (31,32),
and also that the SIRT1 pathway was activated only at the early
phase of drug treatment at sub-lethal concentrations. It has been
reported that biopsies from cancer patients treated with
chemotherapeutic agents also expressed high levels of SIRT1
(33). Consequently, SIRT1 may be
regarded as a potential target for the diagnosis and treatment of
cancer chemotherapy. It was also noted that the inhibition of SIRT1
with NAM sensitized the MCF-7 cells to drug treatment only at low
concentrations, demonstrating that the activated SIRT1 pathway
promoted tumor cell survival through its deacetylase activity. This
conclusion leads to a novel means of improving the clinical
therapeutic effect of tumor chemotherapy.
SIRT1 is an enzyme that catalyzes the deacetylation
of acetyl-lysine residues by a mechanism in which NAD+
is cleaved and O-acetyl ADP-ribose is generated. The reaction
results in the release of NAM, a form of Vitamin B3 that acts as an
end product inhibitor (15). NAM
has been shown to increase radiosensitivity in the course of cancer
radiotherapy. NAM is considered to reduce the occurrence of acute
hypoxia and hence increase tumor blood flow (34,35),
although the precise mechanism of action remains unclear. In the
present study, NAM was used to examine the role of SIRT1 in the
stress response and it was observed that NAM had a synergistic
effect with low concentrations of antitumor agents, thus increasing
chemosensitivity in the course of cancer chemotherapy. The results
also suggested that increased radiosensitivity by NAM may occur via
SIRT1 inhibition.
It has been reported that silencing SIRT1 gene
expression by RNA interference (RNAi) induces growth arrest and
apoptosis in human epithelial cancer cells. By contrast, normal
human epithelial cells and normal human diploid fibroblasts appear
to be refractory to SIRT1 silencing. Therefore, SIRT1 may be
identified as a novel target for the selective killing of cancer
instead of non-cancer epithelial cells (36). In the present study, the SIRT1
deacetylase inhibitor, NAM, induced typical apoptotic features in
the MCF-7 tumor cells. Concentration of 50 mM NAM may be too high
for clinical application, although more sensitive SIRT1 inhibitors,
such as indole and EX527, have been identified as potent and
selective inhibitors of the deacetylase SIRT1 (37).
References
|
1.
|
Imai S, Armstrong CM, Kaeberlein M and
Guarente L: Transcriptional silencing and longevity protein Sir2 is
an NAD-dependent histone deacetylase. Nature. 403:795–800. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Huffman DM, Grizzle WE, Bamman MM, et al:
SIRT1 is significantly elevated in mouse and human prostate cancer.
Cancer Res. 67:6612–6618. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Jang KY, Kim KS, Hwang SH, et al:
Expression and prognostic significance of SIRT1 in ovarian
epithelial tumours. Pathology. 41:366–371. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Cha EJ, Noh SJ, Kwon KS, et al: Expression
of DBC1 and SIRT1 is associated with poor prognosis of gastric
carcinoma. Clin Cancer Res. 15:4453–4459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Stünkel W, Peh BK, Tan YC, et al: Function
of the SIRT1 protein deacetylase in cancer. Biotechnol J.
2:1360–1368. 2007.
|
|
6.
|
Chen J, Zhang B, Wong N, et al: Sirtuin 1
is upregulated in a subset of hepatocellular carcinomas where it is
essential for telomere maintenance and tumor cell growth. Cancer
Res. 71:4138–4149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lin SJ, Defossez PA and Guarente L:
Requirement of NAD and SIR2 for life-span extension by calorie
restriction in Saccharomyces cerevisiae. Science.
289:2126–2128. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Liu T, Liu PY and Marshall GM: The
critical role of the class iii histone deacetylase SIRT1 in cancer.
Cancer Res. 69:1702–1705. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wojcik M, Mac-Marcjanek K and Wozniak LA:
Physiological and pathophysiological functions of SIRT1. Mini Rev
Med Chem. 9:386–394. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Milne JC, Lambert PD, Schenk S, et al:
Small molecule activators of SIRT1 as therapeutics for the
treatment of type 2 diabetes. Nature. 450:712–716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Rodgers JT, Lerin C, Haas W, et al:
Nutrient control of glucose homeostasis through a complex of
PGC-1alpha and SIRT1. Nature. 434:113–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Yamakuchi M, Ferlito M and Lowenstein CJ:
mir-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci
USA. 105:13421–13426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Luo J, Nikolaev AY, Imai S, et al:
Negative control of p53 by Sir2alpha promotes cell survival under
stress. Cell. 107:137–148. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Motta MC, Divecha N, Lemieux M, et al:
Mammalian SIRT1 represses forkhead transcription factors. Cell.
116:551–563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
McBurney MW, Yang X, Jardine K, et al: The
absence of Sir2alpha protein has no effect on global gene silencing
in mouse embryonic stem cells. Mol Cancer Res. 1:402–409.
2003.PubMed/NCBI
|
|
16.
|
Solomon JM, Pasupuleti R, Xu L, et al:
Inhibition of SIRT1 catalytic activity increases p53 acetylation
but does not alter cell survival following DNA damage. Mol Cell
Biol. 26:28–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sauve AA and Schramm VL: Sir2 regulation
by nicotinamide results from switching between base exchange and
deacetylation chemistry. Biochemistry. 42:9249–9256. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Jackson MD, Schmidt MT, Oppenheimer NJ and
Denu JM: Mechanism of nicotinamide inhibition and
transglycosidation by Sir2 histone/protein deacetylases. J Biol
Chem. 278:50985–50998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Borra MT, Langer MR, Slama JT and Denu JM:
Substrate specificity and kinetic mechanism of the sir2 family of
NAD+-dependent histone/protein deacetylases. Biochemistry.
43:9877–9887. 2004.
|
|
20.
|
Bitterman KJ, Anderson RM, Cohen HY, et
al: Inhibition of silencing and accelerated aging by nicotinamide,
a putative negative regulator of yeast sir2 and human SIRT1. J Biol
Chem. 277:45099–45107. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar
|
|
22.
|
Lallemand-Breitenbach V, Zhu J, Chen Z and
de Thé H: Curing APL through PML/RARA degradation by As2O3. Trends
Mol Med. 18:36–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Zhang XW, Yan XJ, Zhou ZR, et al: Arsenic
trioxide controls the fate of the PML-RARalpha oncoprotein by
directly binding PML. Science. 328:240–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Chen G, Wang K, Yang BY, et al:
Synergistic antitumor activity of oridonin and arsenic trioxide on
hepatocellular carcinoma cells. Int J Oncol. 40:139–147.
2012.PubMed/NCBI
|
|
25.
|
Evens AM, Tallman MS and Gartenhaus RB:
The potential of arsenic trioxide in the treatment of malignant
disease: past, present, and future. Leuk Res. 28:891–900. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Wang TH, Wang HS and Soong YK:
Paclitaxel-induced cell death: where the cell cycle and apoptosis
come together. Cancer. 88:2619–2628. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Arcamone F, Cassinelli G, Fantini G, et
al: Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic
from S. Peucetius var. Caesius. Biotechnol Bioeng.
11:1101–1110. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Gewirtz DA: A critical evaluation of the
mechanisms of action proposed for the antitumor effects of the
anthracycline antibiotics adriamycin and daunorubicin. Biochem
Pharmacol. 57:727–741. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Gewirtz DA: Growth arrest and cell death
in the breast tumor cell in response to ionizing radiation and
chemotherapeutic agents which induce DNA damage. Breast Cancer Res
Treat. 62:223–235. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Roninson IB, Broude EV and Chang BD: If
not apoptosis, then what? Treatment-induced senescence and mitotic
catastrophe in tumor cells. Drug Resist Updat. 4:303–313. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Chu F, Chou PM, Zheng X, et al: Control of
multidrug resistance gene mdr1 and cancer resistance to
chemotherapy by the longevity gene sirt1. Cancer Res.
65:10183–10187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Robinson SP, Howe FA, Stubbs M and
Griffiths JR: Effects of nicotinamide and carbogen on tumour
oxygenation, blood flow, energetics and blood glucose levels. Br J
Cancer. 82:2007–2014. 2000.PubMed/NCBI
|
|
35.
|
Hirst DG, Joiner B and Hirst VK: Blood
flow modification by nicotinamide and metoclopramide in mouse
tumours growing in different sites. Br J Cancer. 67:1–6. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Ford J, Jiang M and Milner J:
Cancer-specific functions of SIRT1 enable human epithelial cancer
cell growth and survival. Cancer Res. 65:10457–10463. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Zhao X, Allison D, Condon B, et al: The
2.5 Å crystal structure of the SIRT1 catalytic domain bound to
nicotinamide adenine dinucleotide (NAD+) and an indole (EX527
analogue) reveals a novel mechanism of histone deacetylase
inhibition. J Med Chem. 56:963–969. 2013.
|