Introduction
Pancreatic carcinoma (PC) is an aggressive
malignancy with one of the poorest mortality rates. It is the sixth
leading cause of mortality from malignant disease in China and the
fourth leading cause of cancer-related mortality in the United
States (1–3). The estimated mortality is almost equal
to the estimated incidence, with an overall 5-year survival rate of
<5% (3). Therefore, the
identification of new associated factors and novel therapeutic
targets for PC remains an imperative clinical issue. K-ras, CDKN2A,
p53, DPC4 and 3-phosphoinositide-dependent protein kinase-1 (PDK1)
are known to be mutated or inactivated during PC tumorigenesis, and
mediate other functional and relevant cancer pathways (4–7). PDK1
is a key component in phosphatidylinositol 3-kinase-Akt-mammalian
target of rapamycin (PI3K-Akt-mTOR) signaling, a well-documented
pathway that regulates cancer cell survival and proliferation
(7). PDK1 is a pleckstrin homology
(PH) domain-containing protein that is activated following PI3K
activity, which in turn phosphorylates Akt1 at threonine 308 (or
cognate locations on other isoforms) along with a large variety of
other AGC kinase substrates. Although this kinase has a significant
role in PI3K-Akt-mTOR signaling, the activating mutations of the
gene encoding PDK1 have not been described. The deregulation of
PDK1 is critical to the progression of pancreatic tumors.
MicroRNAs (miRNAs/miRs) are non-coding RNAs that
consist of 21–23 nucleotides and are a recently emerging class of
endogenous negative regulators of gene expression that possess a
remarkable evolutionary conservation (8,9).
miRNAs are believed to modulate gene expression at the
post-transcriptional level (10,11).
These small molecules exert their regulatory effects by
base-pairing to partially complementary mRNA and functioning by two
mechanisms, the degradation of target mRNA transcripts or the
inhibition of mRNA translation (11,12).
miRNAs are also associated with the main phenotypes of numerous
cancer cells (including PC), such as proliferation, invasion and
apoptosis (13–15). Therefore, studying the functions and
mechanisms of miRNAs may lead to new approaches for the
categorization, diagnosis and treatment of human cancers. miR-375
is differentially expressed in various neoplasms (16–18).
The effects of miR-375 may be cell-type specific (16). Few studies of PC have focused on
targeting the clinical and prognostic significance of miR-375.
Our previous study identified that miR-375 is
involved in PC through the suppression of PDK1 (19). However, the detail of how miR-375
regulates PDK1 expression in PC remains unknown.
The present study suggests that miRNAs may play key
roles in tumorigenesis by regulating the expression of genes that
are associated with oncogenic signaling pathways. However, further
studies are required in order to explore and validate whether the
putative target, miR-375, is involved in the regulation of the PDK1
oncogene.
Materials and methods
Tissue samples
PC tissues and their respective adjacent normal
tissues were obtained post-operatively from 44 patients in 2009
from the Department of General Surgery, The First Affiliated
Hospital of Soochow University (Suzhou, Jiangsu, China). Informed
consent was obtained from all patients for their tissues to be used
for scientific research. Approval for the study was obtained from
the Department of General Surgery, The First Affiliated Hospital of
Soochow University. All diagnoses were based on pathological and/or
cytological evidence. The histological features of the specimens
were evaluated by two senior pathologists according to the
classification criteria from the World Health Organization
(20). The tissues were obtained
from the patients prior to chemotherapy or radiation therapy. The
specimens were immediately frozen and stored at −80°C prior to the
microarray and quantitative (q)PCR analyses.
Cell culture
The human PC cell line, Panc-1, was maintained in
DMEM supplemented with 10% FBS. The cells were cultured in an
incubator at 37°C with 5% CO2.
qPCR
Total RNA was extracted from the patients or the
cell line samples using TRIzol (Invitrogen, Carlsbad, CA, USA),
according to the manufacturer’s instructions. The miR-375
expression level was determined by qPCR using Taqman assay kits
(Applied Biosystems, Foster City, CA, USA), with U6 small nuclear
RNA as an internal normalized reference. For the quantification of
PDK1 and β-actin, the extracted RNA was reverse transcribed to cDNA
using an oligo(dT) 12 primer and Superscript II (Invitrogen). The
primers for these genes are summarized in Table I. The relative expression levels of
the primers were measured in triplicate on a Prism 7900 Real-Time
PCR machine (Applied Biosystems), according to the manufacturer’s
instructions.
 | Table IPrimer sequences for qPCR
analysis. |
Table I
Primer sequences for qPCR
analysis.
| Gene | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| PDK1 |
GTGTAGATTAGAGGGATG |
AAGGAATAGTGGGTTAGG |
| β-actin |
CTCCATCCTGGTCTCGCTGT |
GCTGTCACCTTCACCGTTCC |
Vector construction
To generate a miR-375 expression vector (pcDNA
3.1-miR-375) containing a miR-375 precursor, a sequence was
amplified using the following primers: Forward,
5′-CCGCTCGAGCAGATGCGTTCAGGTGAG-3′ and reverse,
5′-CGGAATTCTGGCGGCGGCAGGTGCCTG-3′. The amplified region was cloned
into the pcDNA 3.1+ vector (Genechem, Shanghai, China)
using XbaI and HindIII restriction sites and
confirmed using DNA sequencing, as previously described (21).
The putative miR-375 binding site at the 3′ UTR of
PDK1 was cloned downstream of a CMV promoter-driven firefly
luciferase cassette in a pcDNA 3.1 vector (Genechem). A mutant form
of this luciferase construct was also generated using the PCR-based
overlap-extension procedures, as reported previously (22).
Transfection
Chemically synthesized RNAs, including scramble, an
miR-375 mimic and its inhibitor, were obtained from GenePharma
(Shanghai, China). For transfection, the cells were grown on
24-well culture plates to 70–80% confluence. Following 24 h, the
cells were cotransfected with Renilla luciferase reporter (PRL; 0.1
μg), the previously constructed reporter plasmids (0.5 μg) and
chemically synthesized RNA (0.5 μg) using Lipofectamine 2000
transfection reagent (Invitrogen). Subsequent to being incubated
for 24 h, the cells were harvested using lysis buffer for use in
the luciferase assay. G418 was then used to screen the monoclonal
cells.
Luciferase reporter assay
For the luciferase reporter assay, Panc-1 cells
(3×104) were plated in a 24-well plate and then
cotransfected with 400 ng pcDNA 3.1-miR-375 or pcDNA 3.1 empty
vector, 200 ng wild-type or mutant luciferase construct and 40 ng
PRL-TK (Promega, Madison, WI, USA) using Lipofectamine 2000
(Invitrogen) according to manufacturer’s instructions. The cells
were collected at 48 h post-transfection and analyzed using the
Dual-Luciferase Reporter Assay System (Promega, Fitchburg, WI,
USA). The pRL-TK vector provided the constitutive expression of
Renilla luciferase and was used as an internal control to correct
the differences in the transfection and harvest efficiencies
(23). Each treatment was performed
in triplicate and repeated in three independent experiments.
Western blotting
The treated cells were washed twice with ice-cold
PBS, then directly lysed with 200 μl 2× SDS cell lysis buffer in
each well of a 6-well plate cluster. The lysates were boiled,
centrifuged at 15,680 × g and loaded onto a 12% SDS-PAGE gel. The
samples were electrophoresed for 4 h and transferred to a Millipore
Immobilon transfer membrane (Millipore, Billerica, MA, USA) in
Bio-Rad blot apparatus (Bio-Rad, Hercules, CA, USA). Subsequent to
being blocked with 5% skimmed milk in PBS-Tween-20 for 1 h at room
temperature, the membranes were blotted with a goat polyclonal
anti-hPDK1 primary antibody overnight at 4°C. The membranes were
washed with TBST prior to being incubated with mouse polyclonal
anti-goat secondary antibody linked to horseradish peroxidase
(dilution, 1:2000) for 1 h at room temperature. The membranes were
washed again with TBST and the blot was incubated in a detection
reagent (ECL Advance Western blotting detection kit; Amersham
Bio-science, Freiburg, Germany) and exposed to a Hyperfilm ECL film
(Pierce, Rockford, IL, USA). Anti-PDK1 (Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA) and GAPDH (KangChen Bio-Tech, Shanghai,
China) antibodies were used in the western blot analyses.
Cell proliferation
The cells were seeded in a 96-well plate at a
density of 10,000 cells per well and incubated for 96 h. The in
vitro growth was measured using Cell Counting kit-8 (CCK-8;
Dojindo Laboratories, Kumamoto, Japan). The optical density (OD) at
450 nm was measured using a Microplate Reader (Bio-Rad) and the
proliferation index was calculated as the experimental OD
value/control OD value. A total of three independent experiments
were performed in quadruplicate.
Cell cycle analysis
The cells were washed three times using cold PBS and
then fixed in 70% ethanol in PBS at −20°C for 2 h. Following
fixation, the cells were washed with cold PBS and stained with 1 ml
propidium iodide (PI) staining buffer (MultiScience, Hangzhou,
China), which contained 200 mg/ml RNase A and 50 mg/ml PI, at room
temperature for 30 min in the dark. The analyses were performed on
a FACScan flow cytometer (Becton-Dickinson, Sunnyvale, CA, USA).
The experiments were repeated three times.
Cell apoptosis
The quantification of the apoptotic cells was
performed using the Annexin-V-FITC Apoptosis Detection kit
(Invitrogen), according to the manufacturer’s instructions. Early
apoptotic cells were defined as Annexin-V-positive, PI-negative
cells. The analyses were performed on a FACScan flow cytometer
(Becton-Dickinson). The experiments were repeated three times.
Tumorigenicity assays in nude mice
Six-week-old female BALB/c athymic nude mice were
subcutaneously injected into the right armpit region using
1.5×106 cells in 0.15 ml PBS. The following groups of
mice (n=5 per group) were tested: Group I (miR-375 mimics) was
injected with Panc-1 cells transfected with miR-375 mimics; group
II (Mock) was injected with Panc-1 cells transfected with negative
control; and group III (Vec) was injected with Panc-1 cells alone.
The tumor size was measured every 2 or 3 days using calipers. The
tumor volume was calculated with the following formula: (L ×
W2)/2, where L is the length and W is the width of the
tumor. The mice were sacrificed at four weeks and the weights of
the tumors were measured. All experimental procedures involving
animals were in accordance with the Guide for the Care and Use of
Laboratory Animals (NIH publication no. 80–23, revised 1996) and
were performed according to the ethical guidelines for animal
experiments at the Department of Clinical Immunology, Soochow
University (Suzhou China).
Statistics
The differences between the groups were evaluated
using the Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-375 is a candidate miRNA that
regulates the expression of PDK1
Bioinformatic algorithms (TargetScan 4.2; Whitehead
Institute for Biomedical Research, Cambridge, MA, USA) were used to
identify the candidate miRNAs that targeted the 3′ UTR region of
the PDK1 gene, and to subsequently identify miR-375 for further
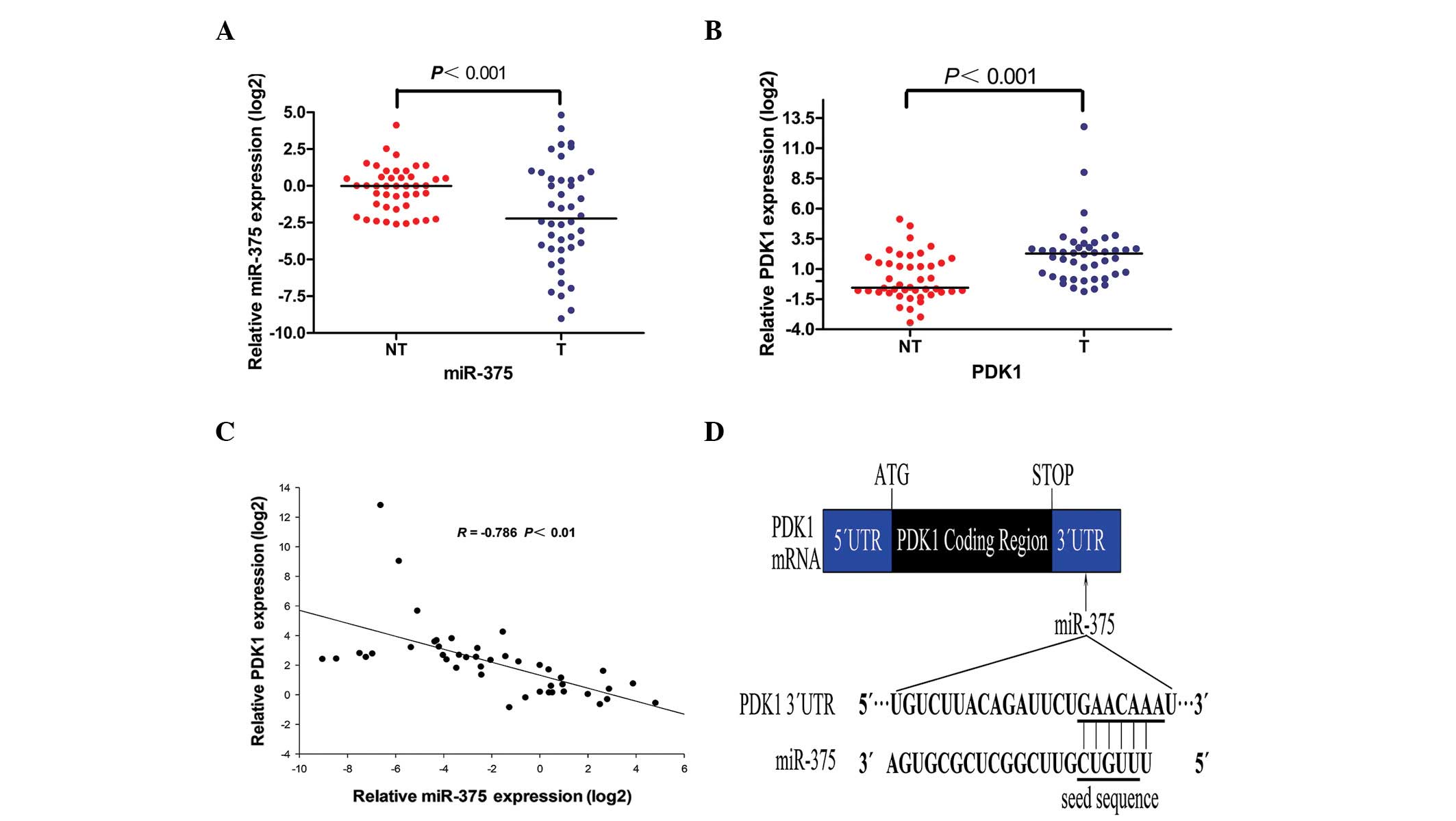
studies (Fig. 1D).
A qPCR analysis of miR-375 and PDK1 was performed in
44 pairs of PC tumor and matched adjacent non-tumor tissues. The
results show that miR-375 was significantly down-regulated in the
PC tumor tissues (Fig. 1A), while
PDK1 was upregulated in the PC tumor tissues (Fig. 1B). PDK1 was inversely correlated
with miR-375 (Fig. 1C), suggesting
that PDK1 is a target of miR-375.
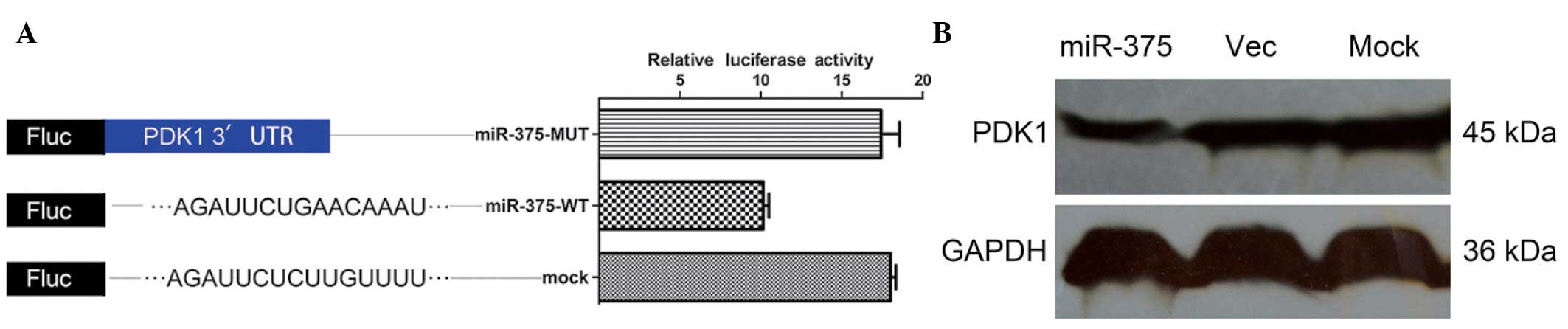
PDK1 is a direct target of miR-375
To confirm that PDK1 is a target of miR-375, a
luciferase reporter assay was performed. The relative luciferase
activity of the reporter that contained the wild-type 3′ UTR was
significantly suppressed when miR-375 was cotransfected (Fig. 2A). In contrast, the luciferase
activity of the mutant reporter was unaffected by the
cotransfection of miR-375 (Fig.
2A), indicating that miR-375 suppressed gene expression using
the miR-375-binding sequence at the 3′ UTR of the PDK1 gene.
The effect of miR-375 on the endogenous expression
of PDK1 was further examined. The ectopic expression of miR-375
caused a decrease in PDK1 protein levels in the Panc-1 cells
(Fig. 2B).
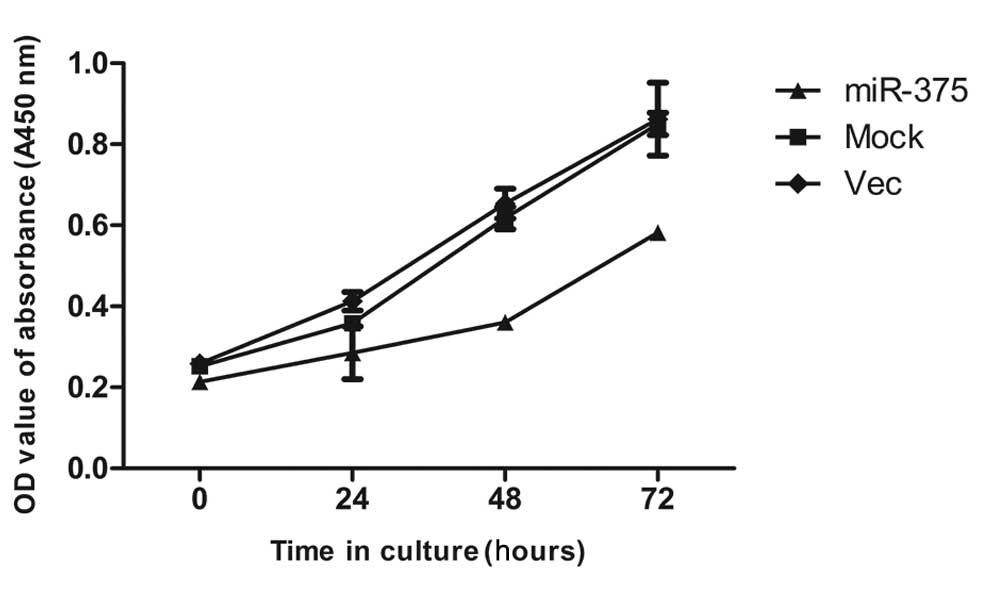
miR-375 suppresses PC cell
proliferation
The significant reduction of miR-375 in the PC
samples and its inhibitory effect on the PDK1 oncogene prompted an
investigation into its possible biological role in cancer cells. To
study the effect of miR-375 on cell proliferation, a CCK-8
proliferation assay was performed.
The results showed that the proliferation of the
pcDNA 3.1-miR-375-transfected cells was slower than the mock and
empty vector-treated cells (Fig.
3).
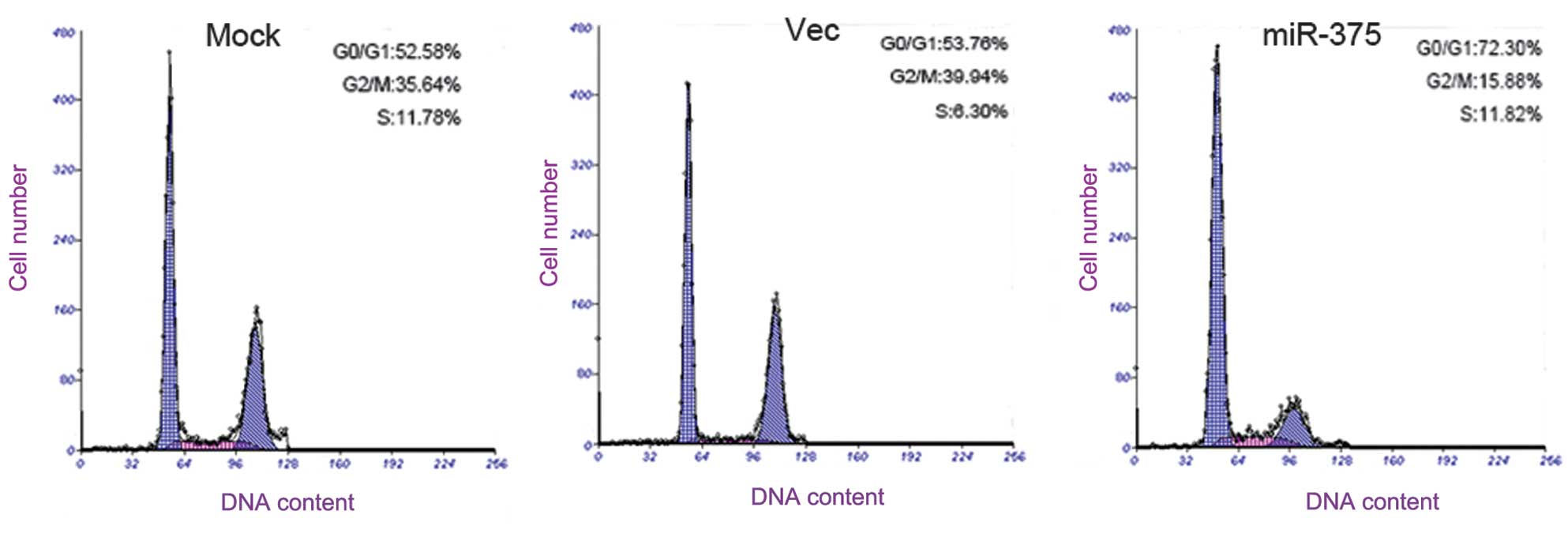
miR-375 arrests the cell cycle at
G0/G1 and induces PC cell apoptosis
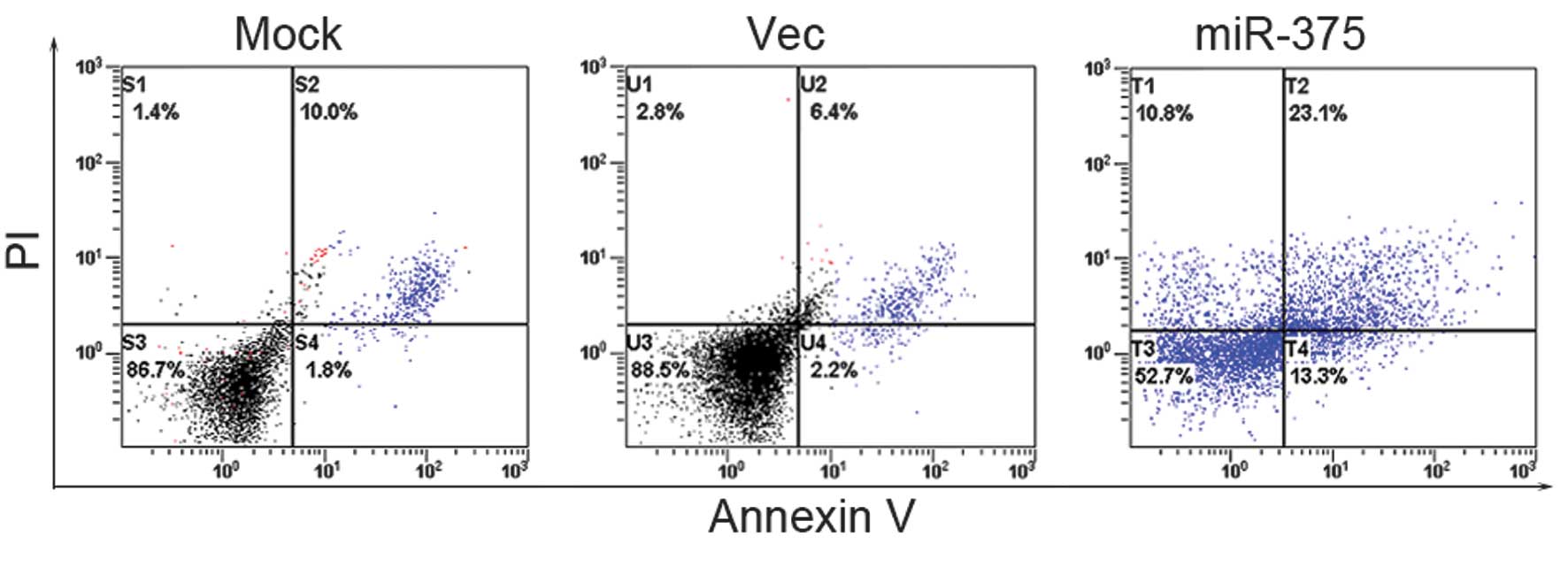
To determine the effect of miR-375 on apoptosis and
the cell cycle of the PC cells, flow cytometry (FCM) was performed.
The results reveal that the Panc-1 cells that were transfected with
the miR-375 mimics had an evident cell cycle arrest at the
G0/G1 phase in contrast with the mock and
empty vector-treated cells (Fig.
4).
The results show that the apoptotic activities of
the pcDNA 3.1-miR-375-transfected cells were higher than that of
the mock and empty vector-treated cells (Fig. 5).
miR-375 suppresses tumorigenicity in
vivo
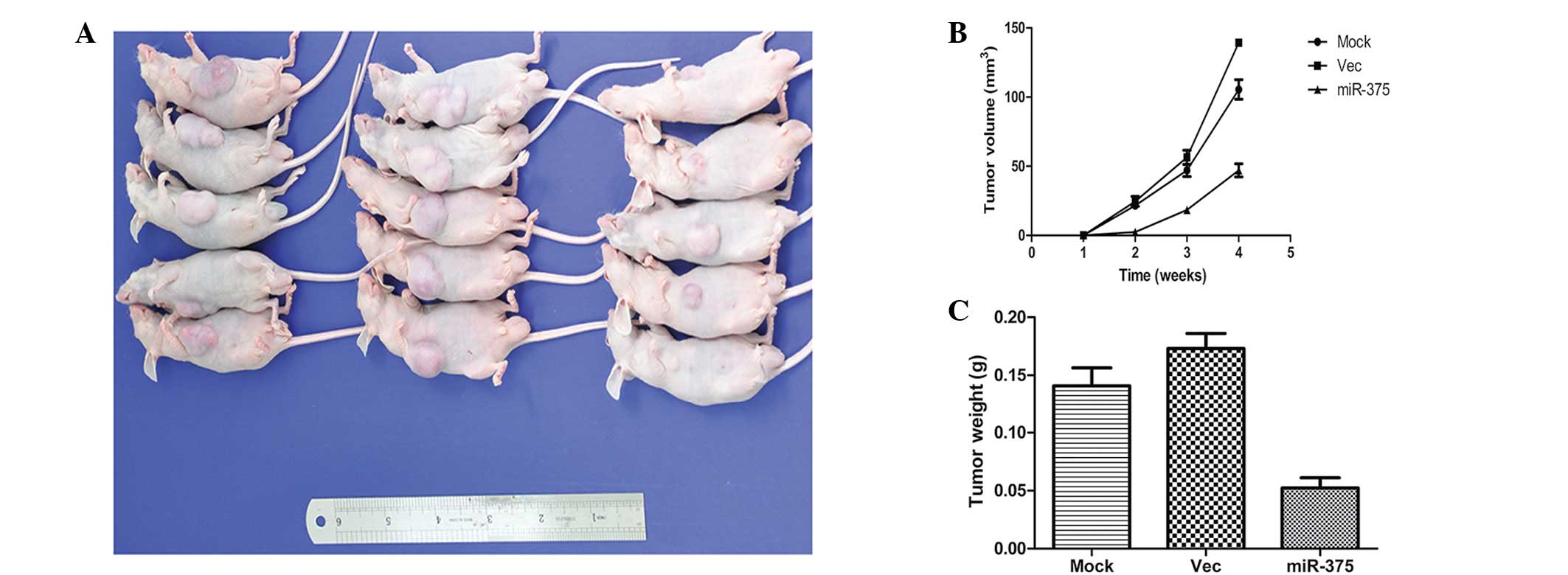
To confirm the aforementioned findings, an in
vivo tumor model was used. miR-375 mimic-transfected Panc-1
cells (miR-375 mimics), negative control-transfected Panc-1 cells
(mock) and Panc-1 cells were injected separately into three groups
of nude mice (n=5 per group). At four weeks post-injection, the
miR-375 mimics group had developed substantially smaller tumors
than the other two groups (Fig.
6A). The tumor volume at the time of death of the mice that
were injected with miR-375 mimic-transfected cells was 60.49±4.59
mm3, whereas the tumor volumes of the mice injected with
mock or Panc-1 cells were 139.28±3.21 mm3 and
105.47±5.89 mm3, respectively (Fig. 6B). Furthermore, the mean tumor
weight at the end of the experiment was markedly lower in the
miR-375 mimics group (0.052±0.013 g) compared with the mock and
Panc-1 groups (0.173±0.028 g and 0.141±0.031 g, respectively;
Fig. 6C).
Discussion
The present study identified that miR-375 was
expressed at a significantly lower level in PC tissues than in the
respective adjacent normal tissues. In the human clinical
specimens, the miR-375 mRNA levels were inversely correlated with
those of PDK1. In addition, miR-375 directly regulated the
expression of PDK1. The PDK1 oncogene was identified to be
negatively regulated by miR-375 at the post-transcriptional level
through a specific target site within the 3′ UTR of the gene. The
overexpression of miR-375 may have resulted in the downregulation
of PDK1. Genomic amplification of PDK1 and the loss of miR-375 may
result in the enhanced expression of the PDK1 oncogene and, in
turn, promote PC development, including the inhibition of cell
proliferation, the induction of apoptosis and cell cycle arrest at
G0/G1. PDK1 is a potent regulator in cell
growth and a previous study supports its oncogenic role in
mammalian cells (7).
Numerous growth factors activate the PI3K pathway,
which in turn phosphorylates phosphatidylinositol-4,5-biphosphate
(PIP2) to generate phosphatidylinositol-3,4,5-triphosphate (PIP3).
An extensively studied signaling event controlled by PIP3 is the
activation of a group of AGC family protein kinases, including
isoforms of protein kinase B (Pkb/Akt) and the ribosomal S6 kinase
(S6K), which play crucial roles in regulating the physiological
processes that are relevant to metabolism, cellular growth,
proliferation and survival (24,25).
PDK1 is a PH domain-containing protein that is activated following
PI3K activity, which in turn phosphorylates Akt1 at threonine 308
(or cognate locations on other isoforms), along with a large
variety of other AGC kinase substrates. Although this kinase has a
significant role in PI3K-Akt-mTOR signaling, the presence of
activating mutations of the gene encoding PDK-1 have not been
described. A study by Westmoreland et al(7) suggested that PDK-1 expression levels
may control the proliferation, survival and growth of developing
pancreatic cells in PC, but this hypothesis has not been fully
tested. PDK1 activation is primarily dependent on cytoplasmic
membrane localization, and is considered to be constitutively
active. Thus, while it is unlikely that there are activating
mutations in the kinase domain, it is possible that
membrane-targeting PDK1 mutations may result in pathway activation
(26).
PDK1 is a gene that has been identified as a direct
target of miR-375 (27) and is a
key component in Akt signaling, a well-documented pathway that
regulates cancer cell survival and proliferation.
The overexpression of miR-375 in PC cells may reduce
cell proliferation and induce cell apoptosis, suggesting a
fundamental role for miR-375 in the development of PC. Accumulating
studies have revealed the significance of miRNAs in regulating the
growth and apoptosis of cancer cells. miR-21 and -155 have been
suggested to function as proto-oncogenes and have been shown to be
overexpressed in several cancers (28). miRNA-146a, which is downregulated in
various cancer types, has been shown to regulate cell proliferation
and apoptosis (29).
A reduction in the expression of miR-375 has been
reported in PC (30),
hepatocellular carcinoma (HCC) (18) and head and neck squamous cell
carcinoma (17). With the exception
of its role in cancer, miR-375 is also a significant regulator in
mammalian pancreatic islet-cell development and in the regulation
of insulin secretion (31), thus
indicating its diverse role in normal physiology. The target genes
of miR-375 may function cooperatively through different cellular
mechanisms. The identification of PDK1 as a target of miR-375
provides new insights into the molecular networks of miR-375.
Further studies are required to investigate the targets of miR-375
that may favor the process of tumorigenesis. An introduction of a
single miRNA may be beneficial in modulating the complex downstream
signals and halting the process of tumorigenesis.
To date, yes-associated protein (YAP) (32), JAK2 (33) and 14-3-3ζ (27) are other genes that have been
identified as direct targets of miR-375. PDK1 has also been
identified as a target of miR-375 in esophageal (34) and gastric cancer (35).
The present study identified that the ectopic
expression of miR-375 may suppress PDK1 mRNA and protein levels,
indicating that miR-375 may also interfere with PI3K-Akt-mTOR
signaling. Therefore, miR-375 is a potential therapeutic target
against the PI3K-Akt-mTOR signaling axis for preventing PC
development and progression.
References
|
1
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar
|
|
2
|
Guo X and Cui Z: Current diagnosis and
treatment of pancreatic cancer in China. Pancreas. 31:13–22. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
4
|
Hingorani SR, Wang L, Multani AS, et al:
Trp53R172H and KrasG12D cooperate to promote chromosomal
instability and widely metastatic pancreatic ductal adenocarcinoma
in mice. Cancer Cell. 7:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bardeesy N, Aguirre AJ, Chu GC, et al:
Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression
of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA.
103:5947–5952. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feldmann G, Beaty R, Hruban RH and Maitra
A: Molecular genetics of pancreatic intraepithelial neoplasia. J
Hepatobiliary Pancreat Surg. 14:224–232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Westmoreland JJ, Wang Q, Bouzaffour M, et
al: Pdk1 activity controls proliferation, survival, and growth of
developing pancreatic cells. Dev Biol. 334:285–298. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bushati N and Cohen SM: microRNA
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007. View Article : Google Scholar
|
|
9
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He L and Hannon GJ: MicroRNAs: small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zaman MS, Chen Y, Deng G, et al: The
functional significance of microRNA-145 in prostate cancer. Br J
Cancer. 103:256–264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu S, Lu Z, Liu C, et al: miRNA-96
suppresses KRAS and functions as a tumor suppressor gene in
pancreatic cancer. Cancer Res. 70:6015–6025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schickel R, Park SM, Murmann AE and Peter
ME: miR-200c regulates induction of apoptosis through CD95 by
targeting FAP-1. Mol Cell. 38:908–915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Szafranska AE, Davison TS, John J, et al:
MicroRNA expression alterations are linked to tumorigenesis and
non-neoplastic processes in pancreatic ductal adenocarcinoma.
Oncogene. 26:4442–4452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Avissar M, Christensen BC, Kelsey KT and
Marsit CJ: MicroRNA expression ratio is predictive of head and neck
squamous cell carcinoma. Clin Cancer Res. 15:2850–2855. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ladeiro Y, Couchy G, Balabaud C, et al:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou J, Song S, Cen J, Zhu D, Li D and
Zhang Z: MicroRNA-375 is downregulated in pancreatic cancer and
inhibits cell proliferation in vitro. Oncol Res. 20:197–203. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hamilton SR and Aaltonen LA: Pathology and
genetics of tumours of the digestive system. World Health
Organization Classification of Tumours. IARC Press; Lyon: 2000
|
|
21
|
Wang XQ, Luk JM, Leung PP, et al:
Alternative mRNA splicing of liver intestine-cadherin in
hepatocellular carcinoma. Clin Cancer Res. 11:483–489.
2005.PubMed/NCBI
|
|
22
|
Wong KF, Luk JM, Cheng RH, et al:
Characterization of two novel LPS-binding sites in leukocyte
integrin betaA domain. FASEB J. 21:3231–3239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu LX, Lee NP, Chan VW, et al: Targeting
cadherin-17 inactivates Wnt signaling and inhibits tumor growth in
liver carcinoma. Hepatology. 50:1453–1463. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kozma SC and Thomas G: Regulation of cell
size in growth, development and human disease: PI3K, PKB and S6K.
Bioessays. 24:65–71. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mora A, Komander D, van Aalten DM and
Alessi DR: PDK1, the master regulator of AGC kinase signal
transduction. Semin Cell Dev Biol. 15:161–170. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Storz P and Toker A:
3′-phosphoinositide-dependent kinase-1 (PDK-1) in PI 3-kinase
signaling. Front Biosci. 7:d886–d902. 2002.
|
|
27
|
El Ouaamari A, Baroukh N, Martens GA, et
al: miR-375 targets 3′-phosphoinositide-dependent protein kinase-1
and regulates glucose-induced biological responses in pancreatic
beta-cells. Diabetes. 57:2708–2717. 2008.
|
|
28
|
Bloomston M, Frankel WL, Petrocca F, et
al: MicroRNA expression patterns to differentiate pancreatic
adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA.
297:1901–1908. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hou Z, Xie L, Yu L, Qian X and Liu B:
MicroRNA-146a is down-regulated in gastric cancer and regulates
cell proliferation and apoptosis. Med Oncol. 29:886–892. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee EJ, Gusev Y, Jiang J, et al:
Expression profiling identifies microRNA signature in pancreatic
cancer. Int J Cancer. 120:1046–1054. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Poy MN, Eliasson L, Krutzfeldt J, et al: A
pancreatic islet-specific microRNA regulates insulin secretion.
Nature. 432:226–230. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu AM, Poon RT and Luk JM: MicroRNA-375
targets Hippo-signaling effector YAP in liver cancer and inhibits
tumor properties. Biochem Biophys Res Commun. 394:623–627. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ding L, Xu Y, Zhang W, et al: MiR-375
frequently downregulated in gastric cancer inhibits cell
proliferation by targeting JAK2. Cell Res. 20:784–793. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Lin R and Li J: Epigenetic silencing
of microRNA-375 regulates PDK1 expression in esophageal cancer. Dig
Dis Sci. 56:2849–2856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsukamoto Y, Nakada C, Noguchi T, et al:
MicroRNA-375 is downregulated in gastric carcinomas and regulates
cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res.
70:2339–2349. 2010. View Article : Google Scholar : PubMed/NCBI
|