Introduction
Ovarian cancer is currently the leading cause of
mortality among gynecological malignant tumors, with epithelial
ovarian cancer (EOC) being the most common, accounting for >85%
of all cases (1). The majority of
ovarian cancers are diagnosed at an advanced stage, mostly due to a
lack of effective screening strategies and difficulties in
obtaining a diagnosis (2). Despite
the progress that has been made in prolonging remission by the
combination of surgical resection and platinum-based chemotherapy,
the overall survival of patients with advanced disease is rarely
>30%. The poor prognosis in the treatment of ovarian cancer is
mainly attributed to chemoresistance (3). Tumor cells may dampen the cytotoxic
effects of anticancer drugs via several mechanisms, including
increased drug efflux, drug inactivation, alteration in the drug
target and increased DNA repair (4,5). As a
result, efforts have been directed towards the development of novel
agents in an attempt to ameliorate the lethality of this
malignancy.
Recent studies on the chemoresistance of ovarian
cancer have indicated that a decreased susceptibility of the cancer
to apoptosis is strongly associated with drug resistance.
Constitutively activated nuclear factor (NF)-κB may be critical in
the development of drug resistance in ovarian cancer cells
(6). NF-κB is known to suppress
apoptosis through the induction of anti-apoptotic proteins,
including Bcl-2 and X-linked inhibitor of apoptosis protein (XIAP),
leading to a resistance to cancer therapy and a poor prognosis
(7–9). Intriguingly, numerous anticancer
drugs, including the DNA-damaging agent cisplatin, are able to
simultaneously stimulate NF-κB activation, as they trigger the cell
death process in neoplasm cells (7,8,10).
Therefore, the inhibition of NF-κB may be useful in increasing the
sensitivity of cells to chemotherapy-dependent apoptosis and
reversing drug resistance in ovarian cancer.
Triptolide (TPL), a purified component extracted
from Tripterygium wilfordii Hook f (TwHf; Lei Gong Teng),
has been identified as the main active element that is responsible
for immunosuppressive and anti-inflammatory properties (11). A number of in vitro and in
vivo studies have revealed that TPL exhibits a wide spectrum of
anticancer effects toward various cancer models (12–17).
However, the underlying molecular mechanisms are complicated and
remain vague. In human anaplastic thyroid carcinoma cells, TPL has
been shown to induce apoptosis through the inhibition of NF-κB in a
p53-independent pathway (13). TPL
has also previously been shown to enhance tumor necrosis factor
(TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis
of lung cancer cells by the inhibition of NF-κB (18). Furthermore, TPL induces the
production of reactive oxygen species (ROS), leading to apoptosis
in human adrenal cancer NCI-H295 cells (16). While certain NF-κB-regulated genes,
including Bcl-2, play a major role in regulating the amount of ROS
in the cell, ROS have various inhibitory and stimulatory roles in
NF-κB signaling (19,20).
The present study aimed to investigate whether TPL
sensitized platinum-resistant SKOV3PT ovarian cancer
cells to apoptosis, along with the molecular signaling pathway
triggered by TPL in platinum-resistant cells. The study further
hypothesized that TPL inactivated the NF-κB pathway through ROS
accumulation, promoting the apoptosis of the SKOV3PT
cells.
Materials and methods
Materials
TPL (Sigma Aldrich Chemical Co., St. Louis, MO, USA)
was dissolved as a stock solution in dimethyl sulfoxide (DMSO) and
freshly diluted in 10 mM culture medium prior to use. Cisplatin,
N-acetyl-L-cysteine (NAC),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI) were obtained from Sigma. 2′,7′-Dichlorodihydrofluorescein
diacetate (H2DCF-DA) was purchased from Calbiochem (San
Diego, CA, USA). The Mitochondrial Isolation kit was bought from
Thermo Scientific (Pierce, Rockford, IL, USA) and the Mitochondrial
Respiratory Chain (MRC) Complexes Activity Assay kits were
purchased from Genmed Scientifics (Shanghai, China). Rabbit
polyclonal anti-Bcl-2 (1:600), rabbit polyclonal anti-NF-κB (p65;
1:400) and goat polyclonal anti-β-actin (1:1000) antibodies were
purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and
rabbit monolonal anti-caspase 3 (1:300) and rabbit monoclonal
anti-XIAP (1:500) antibodies were perchased from Cell Signaling
Technology (San Diego, CA, USA).
Cell culture
The human ovarian carcinoma-derived platinum
resistant SKOV3PT cell line was purchased from the
American Type Culture Collection (Manassas, VA, USA). To maintain
the acquired resistance to cisplatin, the cells were cultured in
RPMI-1640 medium supplemented with fetal bovine serum (10%),
penicillin/streptomycin (100 U/ml) and cisplatin (0.3 μg/ml) in a
5% humidified CO2 atmosphere at 37°C.
Cell viability assay
Cell viability was evaluated using the MTT assay.
Briefly, 1×104 cells/well were seeded in 96-well
microtiter plates. Following the drug treatment, the cells were
incubated with 20 μl MTT (5 mg/ml) for an additional 4 h. The MTT
solution in the medium was discarded and the formazan crystals,
which were formed in the viable cells, were dissolved in 150 μl
DMSO. The optical density of each well was measured at 490 nm using
a Microplate Reader (Molecular Devices, CA, USA).
Apoptosis analysis
Early-stage apoptosis cells that expressed
phosphatidylserine on the outer layer of the cell were detected
using the binding properties of fluoresceinated Annexin V (Annexin
V-FITC). Briefly, the treated cells were harvested and washed twice
with cold phosphate-buffered saline (PBS). The cells were suspended
with a binding buffer and stained with Annexin V-FITC and PI. The
cell mixture was incubated for 15 min at room temperature in the
dark followed by fluorescence-activated cell sorting (FACS)
cater-plus flow cytometry (Becton-Dickinson Co., Heidelberg,
Germany).
ROS detection
The changes in the intracellular ROS levels were
determined using the fluorescent H2DCF-DA probe.
Non-fluorescent H2DCF-DA is cell-permeable, cleaved by
non-specific esterases and oxidized in the presence of ROS to form
fluorescent 2′7′-dichlorofluorescein (DCF). ROS production is
proportional to the fluorescence ratio of the treatment to the
control. The cells were incubated with 10 μM H2DCF-DA
for 20 min at 37°C prior to being harvested and analyzed for
fluorescence intensity using flow cytometry.
Western blotting
Following the treatment of the cells, the nuclear
and cytoplasmic proteins were prepared according to the method
described by Liu et al(21)
and the protein concentrations were measured using a Bicinchoninic
Acid (BCA) Protein Assay kit (Pierce). Equal amounts of proteins
were electrophoresed through denaturing polyacrylamide gels,
transferred onto polyvinylidene difluoride (PVDF) membranes and
probed with primary antibodies against NF-κB (p65), Bcl-2, XIAP and
caspase 3. Subsequent to being washed with TBST, the membranes were
incubated with peroxidase-conjugated secondary antibodies for 1 h.
The blots were detected with an Enhanced Chemiluminescence
Detection kit (Pierce), following the manufacturer's
instructions.
Isolation of mitochondria
The mitochondria were isolated from the cultured
SKOV3PT cells using a Mitochondrial Isolation kit. The
cells were suspended in ice-cold Mito-Cyto isolation buffer and
immediately homogenized. The homogenates were centrifuged at 600 ×
g at 4°C for 10 min. The supernatant was transferred to a new tube
and centrifuged at 11,000 × g at 4°C for 10 min. The pellet was
lysed with Laemmli Buffer (Bio-Rad Laboratories, Hercules, CA, USA)
to extract the mitochondrial protein. The mitochondrial protein
concentration was determined by the BCA Protein Assay kit
(Pierce).
Measurement of mitochondrial complexes I,
II and III activities
The activities of the MRC complexes were determined
using MRC Complexes Activity Assay kits. Mitochondrial complex I
(NADH-ubiquinone oxidoreductase) activity was measured by
monitoring the decrease in NADH absorbance at 340 nm. The activity
of complex I was calculated using the rotenone-sensitive rate and
expressed as μmol/min/mg protein. Complex II (succinate-ubiquinone
oxidoreductase) activity was determined in extracted mitochondria
proteins through the reduction of 2,6-dichloropheno-lindophenol
(DCIP) at 600 nm. The activity of complex II was calculated using
the 2-thenoytrifluoroacetone-sensitive rate and the results were
presented as μmol/min/mg protein. Mitochondrial complex III
(ubiquinol cytochrome-c reductase) activity was measured by
monitoring the reduction of cytochrome-c by ubiquinol at 550 nm and
was expressed as μmol CoQH2/min/mg protein.
Statistical analysis
Each experiment was repeated 3–4 times. The
statistical analysis data were analyzed by one-way ANOVA and are
presented as the mean ± SD. P<0.05 was considered to indicate a
statistically significant difference.
Results
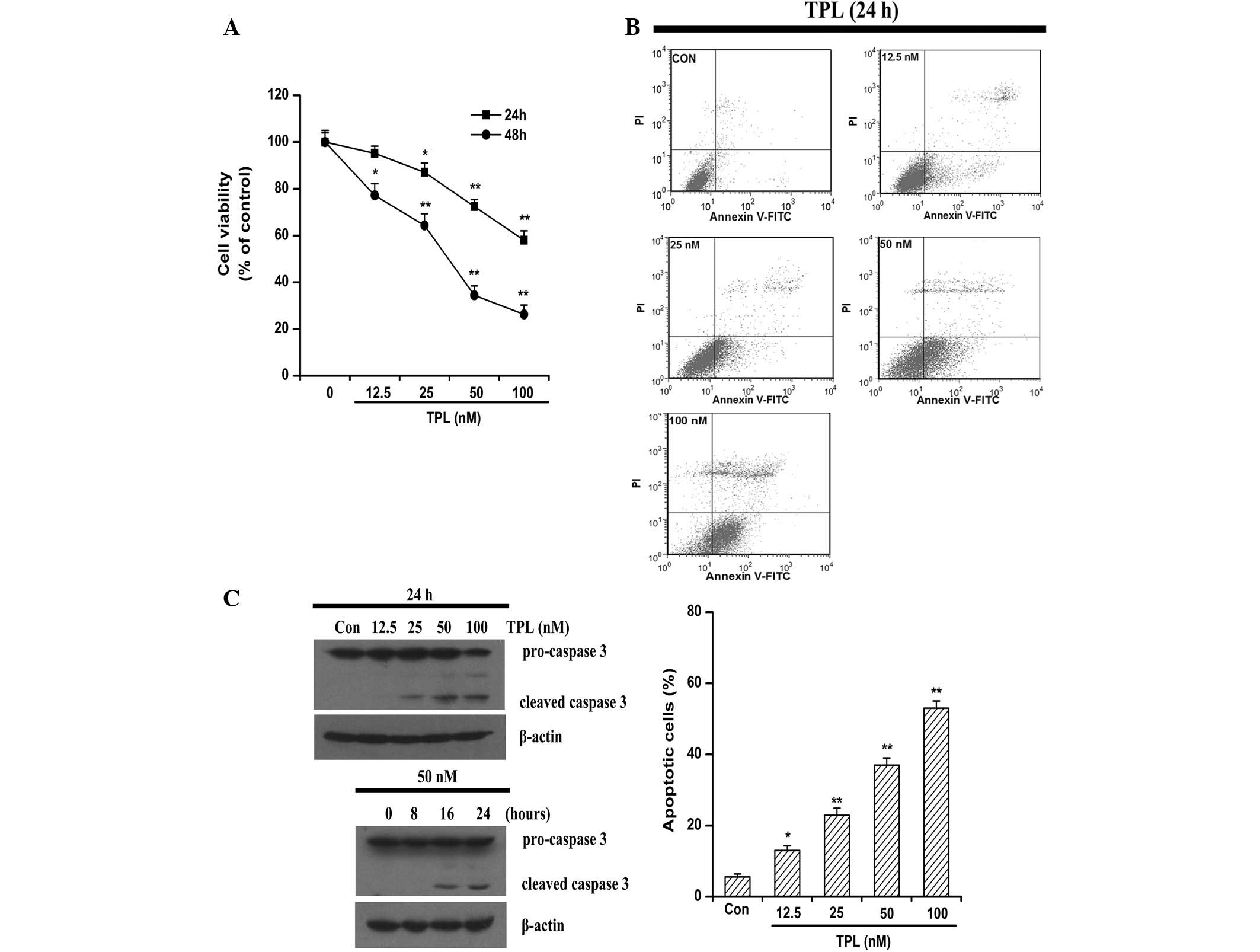
TPL induces apoptosis in the
platinum-resistant SKOV3PT ovarian cancer cell line
The present study evaluated the growth of the
platinum-resistant SKOV3PT ovarian cancer cell line
under the treatment of TPL at various concentrations (0–100 nmol/l)
and time points (24–48 h). The MTT assays revealed that cell
viability was decreased in a dose- and time-dependent manner
following exposure to TPL (Fig.
1A). The 48-h period of TPL exposure inhibited the
proliferation of the SKOV3PT cells with an average
IC50 value of 34.50 nM (Fig.
1A).
To further investigate the cytotoxicity of TPL
against the platinum-resistant SKOV3PT cancer cells, the
cells were subjected to increasing concentrations of TPL and
apoptosis was assessed following 24 h by flow cytometry with
Annexin V/PI staining. As shown in Fig.
1B, TPL treatment mostly induced apoptosis in the
platinum-resistant cells and the proportion of
AnnexinV+/PI− (early stage of apoptosis)
cells increased with the elevated TPL concentrations. Apoptosis is
a tightly regulated, autonomously programmed mechanism that is
finally executed by caspase 3 (22). Caspase 3 is expressed in almost all
types of cells as an inactive pro-enzyme and may be activated by
initiator caspase 8 or caspase 9, which subsequently cleave caspase
3 into two smaller subunits (23).
Compared with the control cells, the caspase 3 activity was
markedly enhanced in the cultures of the cancer cells that were
treated with 25–100 nM TPL for 24 h, as evidenced by a decrease in
density of the pro-form (Fig. 1C).
In a time-dependent experiment, cleavage of caspase 3 was evident
by 16 and 24 h of treatment (Fig.
1C). Taken together, these results indicate that the cytotoxic
effect of TPL is associated with its apoptosis-inducing
activity.
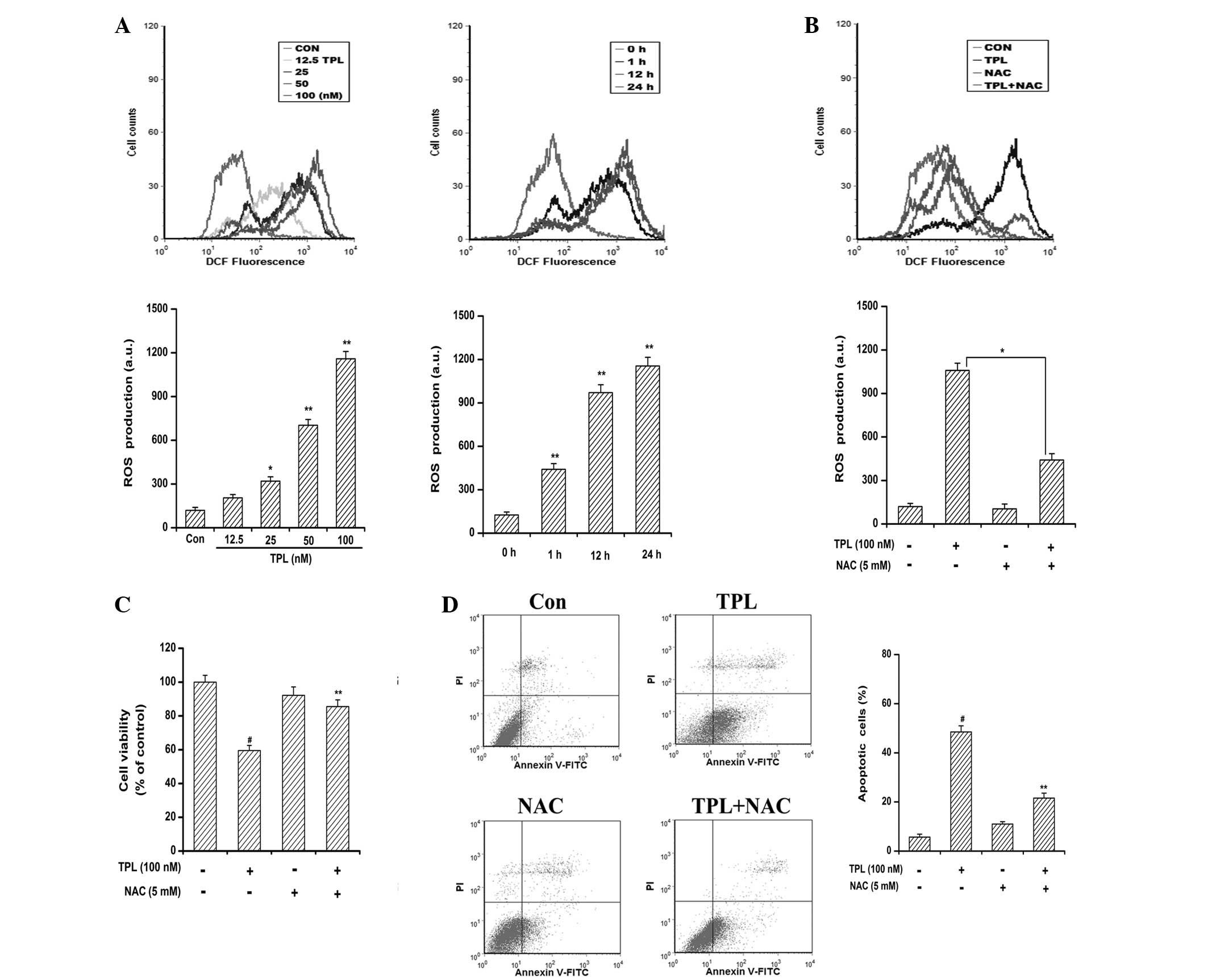
ROS generation is critical for
TPL-induced apoptosis
Numerous anticancer agents exhibit antitumor
activity via the ROS-dependent activation of cancer cell death
(24). It has previously been
reported that elevated intracellular ROS mediates TPL-induced
apoptosis in human adrenal cancer NCI-H295 cells through a
mitochondrial-dependent pathway (16). In the present study, to explore the
involvement of ROS in TPL-induced apoptosis, the generation of ROS
was measured by flow cytometric analysis using H2DCF-DA
dye. As shown in Fig. 2A, TPL
exposure resulted in a time- and concentration-dependent ROS
accumulation in the SKOV3PT cells compared with the
DMSO-treated control cells. Significant ROS generation was observed
when the cells were treated for as little as 1 h and ROS production
was being maintained at a high level by 24 h, indicating a rapid
and sustained generation of ROS in the TPL-treated cells. However,
the production of ROS caused by TPL was greatly reduced by
pre-treatment with NAC due to its ability to elevate intracellular
glutathione to prevent the production of ROS (Fig. 2B). It is of note that the presence
of NAC protected the cells from TPL-induced cytotoxicity (Fig. 2C). Furthermore, the flow cytometric
analyses revealed that the reduction of ROS by NAC attenuated the
number of TPL-induced apoptotic cells from 48.50 to 21.60%
(Fig. 2D). Collectively, these data
suggest that the apoptosis inducing effect of TPL is associated
with ROS generation.
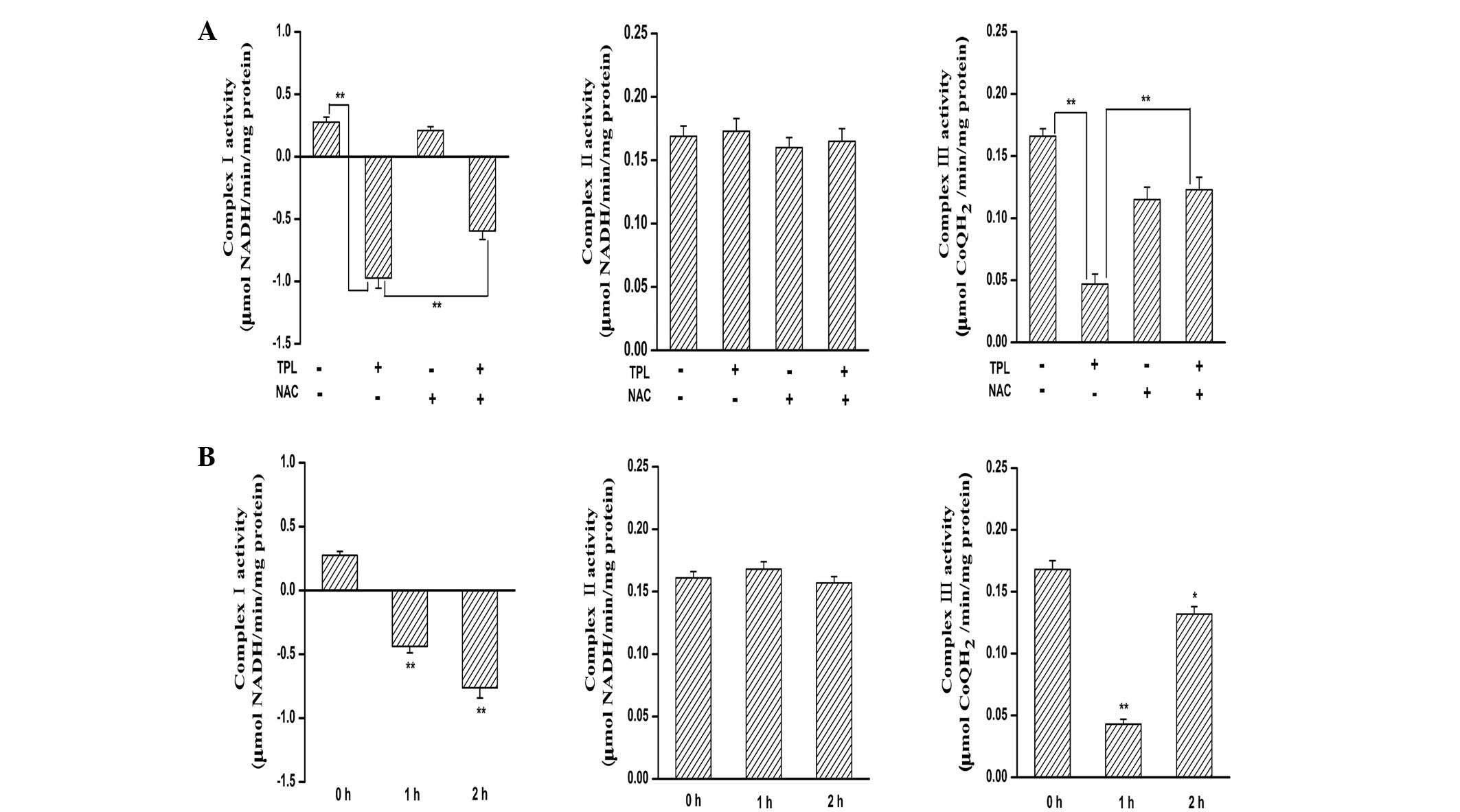
Inhibition of MRC complex I is
responsible for TPL-induced ROS generation
ROS are generated during the electron transport
steps of ATP production via the mitochondrial respiratory chain,
involving auto-oxidation of complexes I, II and III (25). To identify the target of
TPL-mediated ROS generation, the present study determined the
effect of TPL treatment on the activities of MRC enzymes in the
SKOV3PT cells. The activities of MRC complex I and III
decreased in response to the treatment with TPL (Fig. 3A). Pre-treatment with NAC
substantially blocked the decreases in complex III activity by TPL
(Fig. 3A). By contrast, the
inhibitory effect of TPL on complex I was unaffected by NAC
pre-treatment (Fig. 3A). Notably,
complex II activity was not altered by TPL incubation (Fig. 3A). These results indicate that
mitochondrial complex I appears to be responsible for ROS
generation triggered by TPL. Additionally, the activity of MRC
complex I decreased as early as 1 h following TPL exposure
(Fig. 3B), which was consistent
with the kinetics of ROS generation. However, the same batch and
same concentration of TPL did not inhibit complex II and III
activities, even following a 2-h treatment period (Fig. 3B). Accordingly, these data further
lead us to speculate that TPL may cause ROS generation, at least in
part, by inhibiting mitochondrial complex I activity.
TPL treatment causes ROS-dependent
suppression of NF-κB activation and cleavage of pro-caspase 3
NF-κB has been strongly implicated in cell
proliferation, survival and chemoresistance in multiple tumors
(7,8). In normal cells, NF-κB predominantly
resides in the cytosol due to the inhibitory protein, IκBα, but it
is translocated to the nucleus upon growth or survival stimulation
(7). Therefore, the present study
examined the effect of TPL treatment on the cellular localization
of NF-κB (p65). As observed in Fig.
4A, the SKOV3PT cells expressed substantial levels
of nuclear p65 protein, implying that NF-κB is constitutively
activated in these cells. Compared with the control cells, the
nuclear content of p65 protein was significantly decreased in the
TPL-treatment cells (Fig. 4A). The
role of NF-κB in chemotherapeutic drug resistance has been
associated with the induction of survival signals through the
upregulation of anti-apoptotic proteins, including Bcl-2 and XIAP
(8,9). As expected, the expression levels of
Bcl-2 and XIAP were also decreased under TPL treatment (Fig. 4A). Notably, the TPL-induced
reduction in the expression of the nuclear p65 protein and
cytoplasmic Bcl-2 and XIAP proteins was inhibited by pre-treatment
with NAC compared with TPL treatment alone (Fig. 4B). The effect of NAC on the
activation of caspase 3 induced by TPL was subsequently examined.
When the cells were treated in the presence of NAC, the cleavage of
pro-caspase 3 induced by TPL was evidently suppressed (Fig. 4B). These results suggest that the
inhibition of the NF-κB pathway is associated with the increased
levels of intracellular ROS induced by TPL during apoptosis of
platinum-resistant ovarian cancer cells.
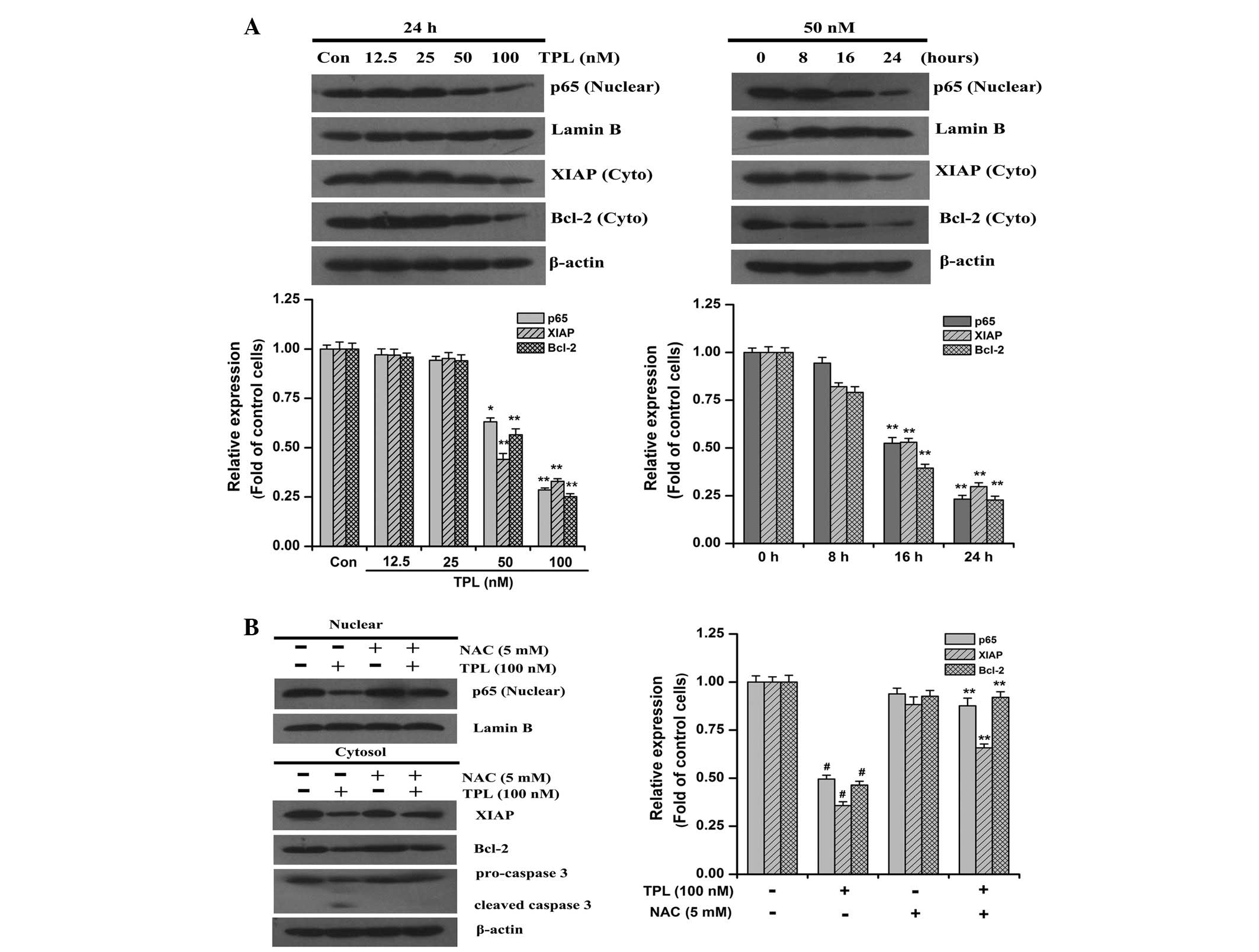 | Figure 4ROS-mediated TPL-induced
downregulation of NF-κB/p65, Bcl-2, XIAP and cleavage of
pro-caspase 3 in SKOV3PT cells. (A) Western blot
analysis of NF-κB/nuclear p65, XIAP and Bcl-2 following exposure to
the indicated concentrations of TPL (12.5–100 nM) for 24 h or at
various time points with 50 nM TPL treatment. (B) Western blot
analysis of NF-κB/nuclear p65, XIAP, Bcl-2 and caspase 3 in
SKOV3PT cells that were pre-treated with and without 5
mM NAC for 1 h followed by 100 nM TPL for 24 h. Lamin B and β-Actin
served as nuclear and cytosolic internal controls, respectively.
The relative levels of protein expression are shown with the
densitometric analysis and the values are expressed as the mean ±
SD of three experiments. (A) *P<0.05 and
**P<0.01 vs. the control. (B) #P<0.01
vs. the control group and **P<0.01 vs. TPL without
NAC. ROS, reactive oxygen species; TPL, triptolide; NF-κB, nuclear
factor-κB; XIAP, X-linked inhibitor of apoptosis protein; NAC, NAC,
N-acetyl-L-cysteine. |
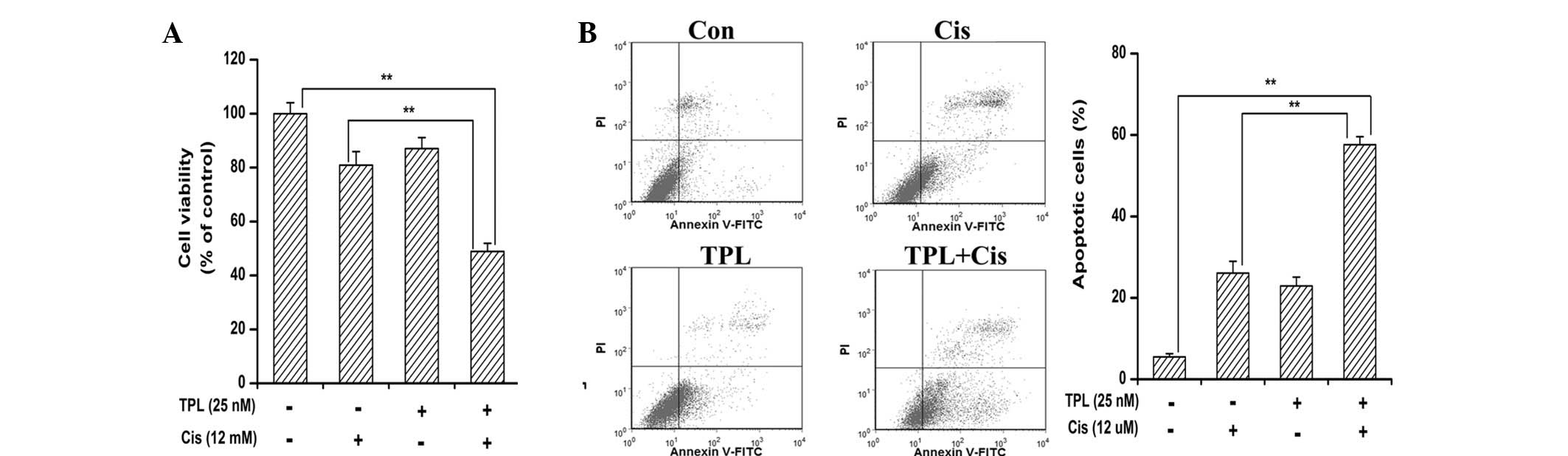
TPL synergistically enhances
cisplatin-induced cytotoxicity in platinum-resistant cells
As the delivery of lower dose agents result in lower
toxicity and an increase in patient tolerance, strategies using
novel effectively safe agents or drug combinations are being
increasingly investigated for overcoming chemoresistance (26,27).
The present study tested whether low doses of the two drugs in
combination were able to exert a synergistic anticancer effect
in vitro compared with TPL or cisplatin alone. A dose of 12
μM cisplatin, which was the IC50 of cisplatin on the
parental SKOV3PT cells (28), had a minimal effect on cell
viability in the platinum-resistant SKOV3PT cells
(Fig. 5A). This indicated that the
SKOV3PT cells were relatively resistant to cisplatin. In
combination with 25 nM TPL, the inhibition rate was rapidly
increased to 51.10% (Fig. 5A). TPL
alone (25 nM) in the previous data revealed 12.95% cell death
(Fig. 1A). The combination index
(CI) of the combination was <1, suggesting that the
antiproliferative effect of the combination was synergistic rather
than additive. These data demonstrate that TPL is able to sensitize
platinum-resistant SKOV3PT cells to cisplatin.
Additionally, the Annexin V apoptosis assay revealed that TPL
enhanced the apoptotic effect of cisplatin from 26.07 to 57.60%
(Fig. 5B). These observations
demonstrate that TPL combined with cisplatin exhibits synergistic
effects against platinum-resistant cells.
Discussion
Although first-line platinum-based chemotherapy
following an apparent curative resection has improved survival
length, severe adverse side-effects and drug resistance have
emerged as the major impediments to effective ovarian cancer
therapy (29). Thus, novel
strategies involving less toxic agents that are able to either
enhance the antitumor effects of cisplatin or overcome
chemoresistance to the drug are highly desirable.
The pleiotropic anticancer activities of TPL have
attracted a great deal of research interest. TPL has been shown to
possess the capacity to inhibit proliferation and induce apoptosis
of various cancer cell lines in vitro and in
vivo(12–17). Notably, TPL has also been identified
to be effective in the induction of apoptosis in drug-resistant
multiple myeloma (30) and cervical
cancer (31) cells. Therefore, the
present study investigated whether TPL treatment was able to
exhibit a cytotoxic effect on platinum-resistant ovarian cancer
cells. The results demonstrated that TPL reduced the growth of the
platinum-resistant ovarian cancer cells by inducing apoptosis,
evidenced by the externalization of membrane-bound
phosphatidylserine and the cleavage of caspase 3. The results also
showed that the addition of a low concentration of TPL greatly
increased the cytotoxicity of cisplatin against the
SKOV3PT cells, which is consistent with previous studies
(30,31).
The intracellular redox status, regulated by the
production of ROS, greatly contributes to the regulation of cell
survival and death (32). Oxidative
stress is the condition arising from an imbalance between the
production of intracellular ROS and the ability of cells to defend
themselves against them (33).
Although cancer cells become well adapted to persistent intrinsic
oxidative stress, a further increase in ROS above the toxic
threshold level may result in cell death (34). It is noteworthy that numerous
commonly used chemotherapy agents, including cisplatin and
etoposide, may trigger the ROS-dependent activation of apoptotic
cell death (35,36). However, continuous cisplatin
treatment may reduce cellular ROS levels and cancer cells
containing reduced ROS may become drug resistant cells (37). Furthermore, an elevation of the
cellular ROS level by exogenous ROS generation in combination with
cisplatin resensitizes drug-resistant cancer cells (37). Several studies have attributed ROS
generation to the pro-apoptotic effect of TPL in various cell types
(16,38,39),
which is in agreement with the findings of the present study.
Growing evidence supports a role for ROS in the
modulation of signaling pathways, which are necessary for cell
proliferation, differentiation and cancer metastasis (34,40).
However, prolonged and high levels of ROS may be indicative of the
stimulation of a cellular death signal via activating cell surface
death receptors or acting directly on the mitochondria (41). Although a possible contribution of
ROS has been observed in the apoptotic response to TPL, the
mechanism by which TPL treatment causes ROS generation is unclear.
Mitochondria are a major source of cellular ROS, particularly
through electron leakage from the respiratory complexes (25). The inhibition of MRC complex
activity is capable of leaking electrons to react with molecular
oxygen, resulting in the formation of ROS (42,43).
The present study demonstrated that ROS generation by TPL in
platinum-resistant cancer cells occurs through MRC complex I. The
activity of MRC complex I decreased following TPL treatment and NAC
did not reverse the inhibition (Fig.
3A). Furthermore, the pattern of TPL-mediated ROS generation
closely mirrored the inhibition of complex I activity (Fig. 3B). These results are consistent with
a previous study in which celestrol induced ROS-dependent
cytotoxicity by targeting MRC complex I (43). The precise mechanism of the
TPL-mediated inhibition of complex I activity remains to be
elucidated.
NF-κB signaling is one of the major pathways
responsible for the platinum resistance of ovarian cancer, as
reflected by the fact that its basal activity is significantly
increased in platinum-resistant Caov-3 cells compared with A2780
platinum-sensitive cells (44). In
cisplatin-resistant Caov-3 ovarian cancer cells, the inhibition of
NF-κB activity by treatment with specific NF-κB nuclear
translocation inhibitors (SN-50) or by the transfection of p50
ΔNLS, which lacks the nuclear localization signal domain, increased
the efficacy of cisplatin-induced apoptosis (44). TPL has been identified as a novel
NF-κB inhibitor and has been shown to increase the efficacy of
5-fluorouracil (FU) and TNF in cancer cells through the inhibition
of NF-κB activity (38,45). The present study demonstrated that
the SKOV3PT cells contained substantial levels of
nuclear p65 protein, which implies that substantial NF-κB activity
may confer survival in these cells. The data from this study
revealed that TPL blocked the transactivation of p65. Therefore the
inhibition of NF-κB may account for TPL-induced cell death.
Previous studies have shown that acquired cisplatin resistance in
ovarian cancer is correlated with an increased expression of Bcl-2
and XIAP (46,47), which are regulated by NF-κB. The
present study observed that TPL treatment led to a reduction in the
expression of Bcl-2 and XIAP, which is consistent with a previous
study (45). Accordingly, the
inactivation of the NF-κB survival pathway may be a significant
molecular mechanism contributing to the cisplatin resistance
reversing effect of TPL.
NF-κB is a redox-sensitive transcription factor
(48). NF-κB may be activated by
ROS, resulting in the transcriptional activation of a variety of
genes that are involved in cell transformation, proliferation and
angiogenesis (19,49). Notably, the antioxidant, NAC,
effectively blocked the intracellular ROS induced by TPL and
simultaneously suppressed the reduction of p65, Bcl-2 and XIAP
expression and the activation of caspase 3. These data indicate
that the apoptotic effect of TPL on cancer cells through the
accumulation of intracellular ROS, may function upstream of NF-κB
and caspase 3. It is highly likely that the presence of oxidative
stress may be decisive for the ability of TPL to inhibit NF-κB in
the present study. Korn et al(50) reported that
H2O2 was capable of inhibiting TNF-induced
NF-κB activation in lung epithelial cells by the reduction in
inhibitor of NF-κB kinase (IKK)-β activity through the oxidation of
cysteine residues in the IKK complex. IKKβ inactivation through the
oxidation of IKKβ on cysteine 179 has also been shown in arsenite
treatment, leading to a reduction in NF-κB signaling (51). Further studies are required to
experimentally explore these possibilities.
Altogether, the present study offers the first
evidence that ROS that are produced in response to TPL treatment
via a marked inhibition of mitochondrial complex I lead to NF-κB
inactivation and initiate caspase 3-mediated apoptosis in
platinum-resistant cancer cells. Furthermore, TPL acted
co-operatively with cisplatin to induce apoptosis in the
platinum-resistant cells. Further in vivo experiments may
aid in the confirmation of the therapeutic efficacy of this agent
for female patients with platinum-resistant ovarian cancer.
Acknowledgements
This study was supported by the Natural Scientific
Foundation of China (grant nos. 81060022 and 81260491).
References
|
1
|
Cannistra SA: Cancer of the ovary. N Engl
J Med. 351:2519–2529. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shepherd JE: Current strategies for
prevention, detection, and treatment of ovarian cancer. J Am Pharm
Assoc (Wash). 40:392–401. 2000.PubMed/NCBI
|
|
3
|
Agarwal R and Kaye SB: Ovarian cancer:
strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brabec V and Kasparkova J: Modifications
of DNA by platinum complexes. Relation to resistance of tumors to
platinum antitumor drugs. Drug Resist Updat. 8:131–146. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
White KL, Rider DN, Kalli KR, Knutson KL,
Jarvik GP and Goode EL: Genomics of the NF-κB signaling pathway:
hypothesized role in ovarian cancer. Cancer Causes Control.
22:785–801. 2011.
|
|
7
|
Nakanishi C and Toi M: Nuclear
factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat
Rev Cancer. 5:297–309. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li F and Sethi G: Targeting transcription
factor NF-kappaB to overcome chemoresistance and radioresistance in
cancer therapy. Biochim Biophys Acta. 1805:167–180. 2010.PubMed/NCBI
|
|
9
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yeh PY, Chuang SE, Yeh KH, Song YC, Ea CK
and Cheng AL: Increase of the resistance of human cervical
carcinoma cells to cisplatin by inhibition of the MEK to ERK
signaling pathway partly via enhancement of anticancer drug-induced
NF kappa B activation. Biochem Pharmacol. 63:1423–1430. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen BJ: Triptolide, a novel
immunosuppressive and anti-inflammatory agent purified from a
Chinese herb Tripterygium wilfordii Hook F. Leuk Lymphoma.
42:253–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen YW, Lin GJ, Chia WT, Lin CK, Chuang
YP and Sytwu HK: Triptolide exerts anti-tumor effect on oral cancer
and KB cells in vitro and in vivo. Oral Oncol. 45:562–568. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu W, Hu H, Qiu P and Yan G: Triptolide
induces apoptosis in human anaplastic thyroid carcinoma cells by a
p53-independent but NF-kappaB-related mechanism. Oncol Rep.
22:1397–1401. 2009.PubMed/NCBI
|
|
14
|
Li H, Takai N, Yuge A, et al: Novel target
genes responsive to the anti-growth activity of triptolide in
endometrial and ovarian cancer cells. Cancer Lett. 297:198–206.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim MJ, Lee TH, Kim SH, Choi YJ, Heo J and
Kim YH: Triptolide inactivates Akt and induces caspase-dependent
death in cervical cancer cells via the mitochondrial pathway. Int J
Oncol. 37:1177–1185. 2010.PubMed/NCBI
|
|
16
|
Wu PP, Liu KC, Huang WW, et al: Triptolide
induces apoptosis in human adrenal cancer NCI-H295 cells through a
mitochondrial-dependent pathway. Oncol Rep. 25:551–557.
2011.PubMed/NCBI
|
|
17
|
Antonoff MB, Chugh R, Borja-Cacho D, et
al: Triptolide therapy for neuroblastoma decreases cell viability
in vitro and inhibits tumor growth in vivo. Surgery. 146:282–290.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee KY, Park JS, Jee YK and Rosen GD:
Triptolide sensitizes lung cancer cells to TNF-related
apoptosis-inducing ligand (TRAIL)-induced apoptosis by inhibition
of NF-kappaB activation. Exp Mol Med. 34:462–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morgan MJ and Liu ZG: Crosstalk of
reactive oxygen species and NF-kappaB signaling. Cell Res.
21:103–115. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kane DJ, Sarafian TA, Anton R, et al:
Bcl-2 inhibition of neural death: decreased generation of reactive
oxygen species. Science. 262:1274–1277. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu GH, Wang SR, Wang B and Kong BH:
Inhibition of nuclear factor-kappaB by an antioxidant enhances
paclitaxel sensitivity in ovarian carcinoma cell line. Int J
Gynecol Cancer. 16:1777–1782. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cohen GM: Caspases: the executioners of
apoptosis. Biochem J. 326:1–16. 1997.
|
|
23
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fang J, Nakamura H and Iyer AK:
Tumor-targeted induction of oxystress for cancer therapy. J Drug
Target. 15:475–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adam-Vizi V and Chinopoulos C:
Bioenergetics and the formation of mitochondrial reactive oxygen
species. Trends Pharmacol Sci. 27:639–645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silasi DA, Alvero AB, Rutherford TJ, Brown
D and Mor G: Phenoxodiol: pharmacology and clinical experience in
cancer monotherapy and in combination with chemotherapeutic drugs.
Expert Opin Pharmacother. 10:1059–1067. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sung B, Kunnumakkara AB, Sethi G, Anand P,
Guha S and Aggarwal BB: Curcumin circumvents chemoresistance in
vitro and potentiates the effect of thalidomide and bortezomib
against human multiple myeloma in nude mice model. Mol Cancer Ther.
8:959–970. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang YI, Kim JH, Lee KT and Choi JH:
Costunolide induces apoptosis in platinum-resistant human ovarian
cancer cells by generating reactive oxygen species. Gynecol Oncol.
123:588–596. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Berkenblit A and Cannistra SA: Advances in
the management of epithelial ovarian cancer. J Reprod Med.
50:426–438. 2005.
|
|
30
|
Yang M, Huang J, Pan HZ and Jin J:
Triptolide overcomes dexamethasone resistance and enhanced
PS-341-induced apoptosis via PI3k/Akt/NF-kappaB pathways in human
multiple myeloma cells. Int J Mol Med. 22:489–496. 2008.PubMed/NCBI
|
|
31
|
Chen YW, Lin GJ, Chuang YP, et al:
Triptolide circumvents drug-resistant effect and enhances
5-fluorouracil antitumor effect on KB cells. Anticancer Drugs.
21:502–513. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hampton MB and Orrenius S: Redox
regulation of apoptotic cell death. Biofactors. 8:1–5. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Toyokuni S, Okamoto K, Yodoi J and Hiai H:
Persistent oxidative stress in cancer. FEBS Lett. 358:1–3. 1995.
View Article : Google Scholar
|
|
34
|
Fruehauf JP and Meyskens FL Jr: Reactive
oxygen species: a breath of life or death? Clin Cancer Res.
13:789–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maiti AK: Genetic determinants of
oxidative stress-mediated sensitization of drug-resistant cancer
cells. Int J Cancer. 130:1–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kurosu T, Fukuda T, Miki T and Miura O:
BCL6 overexpression prevents increase in reactive oxygen species
and inhibits apoptosis induced by chemotherapeutic reagents in
B-cell lymphoma cells. Oncogene. 22:4459–4468. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maiti AK: Gene network analysis of
oxidative stress-mediated drug sensitivity in resistant ovarian
carcinoma cells. Pharmacogenomics J. 10:94–104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu B, Guo X, Mathew S, et al: Triptolide
simultaneously induces reactive oxygen species, inhibits NF-kappaB
activity and sensitizes 5-fluorouracil in colorectal cancer cell
lines. Cancer Lett. 291:200–208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bao X, Cui J, Wu Y, et al: The roles of
endogenous reactive oxygen species and nitric oxide in
triptolide-induced apoptotic cell death in macrophages. J Mol Med
(Berl). 85:85–98. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lau AT, Wang Y and Chiu JF: Reactive
oxygen species: current knowledge and applications in cancer
research and therapeutic. J Cell Biochem. 104:657–667. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Engel RH and Evens AM: Oxidative stress
and apoptosis: a new treatment paradigm in cancer. Front Biosci.
11:300–312. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dias N and Bailly C: Drugs targeting
mitochondrial functions to control tumor cell growth. Biochem
Pharmacol. 70:1–12. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen G, Zhang X, Zhao M, et al: Celastrol
targets mitochondrial respiratory chain complex I to induce
reactive oxygen species-dependent cytotoxicity in tumor cells. BMC
Cancer. 11:1702011. View Article : Google Scholar
|
|
44
|
Mabuchi S, Ohmichi M, Nishio Y, et al:
Inhibition of NFkappaB increases the efficacy of cisplatin in in
vitro and in vivo ovarian cancer models. J Biol Chem.
279:23477–23485. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Park B, Sung B, Yadav VR, Chaturvedi MM
and Aggarwal BB: Triptolide, histone acetyltransferase inhibitor,
suppresses growth and chemosensitizes leukemic cells through
inhibition of gene expression regulated by
TNF-TNFR1-TRADD-TRAF2-NIK-TAK1-IKK pathway. Biochem Pharmacol.
82:1134–1144. 2011. View Article : Google Scholar
|
|
46
|
Wang J, Zhou JY, Zhang L and Wu GS:
Involvement of MKP-1 and Bcl-2 in acquired cisplatin resistance in
ovarian cancer cells. Cell Cycle. 8:3191–3198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mansouri A, Zhang Q, Ridgway LD, Tian L
and Claret FX: Cisplatin resistance in an ovarian carcinoma is
associated with a defect in programmed cell death control through
XIAP regulation. Oncol Res. 13:399–404. 2003.PubMed/NCBI
|
|
48
|
Schreck R, Rieber P and Baeuerle PA:
Reactive oxygen intermediates as apparently widely used messengers
in the activation of the NF-kappa B transcription factor and HIV-1.
EMBO J. 10:2247–2258. 1991.PubMed/NCBI
|
|
49
|
Van Waes C: Nuclear factor-kappaB in
development, prevention, and therapy of cancer. Clin Cancer Res.
13:1076–1082. 2007.PubMed/NCBI
|
|
50
|
Korn SH, Wouters EF, Vos N and
Janssen-Heininger YM: Cytokine-induced activation of nuclear
factor-kappa B is inhibited by hydrogen peroxide through oxidative
inactivation of IkappaB kinase. J Biol Chem. 276:35693–35700. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kapahi P, Takahashi T, Natoli G, et al:
Inhibition of NF-kappa B activation by arsenite through reaction
with a critical cysteine in the activation loop of Ikappa B kinase.
J Biol Chem. 275:36062–36066. 2000. View Article : Google Scholar : PubMed/NCBI
|