Introduction
Neurocytomas are rare tumors hypothesized to
originate from bipotent progenitor cells, with the potential for
neuronal and glial differentiation (1,2).
Extraventricular neurocytomas (EVNs) are rare neuronal tumors
included in the definition of neoplasms in the 2007 World Health
Organization (WHO) classification of tumors of the central nervous
system (3). EVNs tend to be large,
well-circumscribed lesions located in the cerebral hemispheres that
are commonly identified in the frontal and parietal lobes. However,
EVNs have been located in the thalamus, cerebellum, pineal region
and even in the spinal cord (4–6).
Unlike the usual intraventricular central neurocytomas (CN),
typical EVNs exhibit a wide spectrum of morphologies, including the
growth of monotonous neurocytes in sheets, clusters, ribbons or
rosettes and neurophils dispersed in broad zones (7). Although EVNs exhibit histologically
uniform round cells, i.e. a clear cell morphology with neuronal
differentiation, a clear cell morphology is also a classic feature
of oligodendroglioma. Thus, the morphological overlap of these
tumors often creates diagnostic difficulties (8,9).
Moreover, increasing evidence of oligodendrogliomas with neuronal
or neurocytic differentiation makes the distinction of EVNs
increasingly difficult despite various immunohistochemistry methods
of examination (10). Familiarity
with the histopathological features of neurocytoma is a necessity
for pathologists and surgeons in order to avoid such a misdiagnosis
and to provide optimal therapy. While a small case series of EVNs
in adults has been previously reported, EVNs in the pediatric
population are extremely rare (11). To aid the clarification of the
spectrum of such lesions and their biological behavior in the
pediatric population, the present case report presents the
clinicopathological features of an EVN in a 2-year-old female. The
patient was admitted to hospital with nausea and vomiting that had
lasted for five days and a large right frontal lobe mass that was
later determined to be an EVN. In addition, the current study
presents an analysis of the imaging features, histology, treatment
and prognosis of these reported rare lesions. Written informed
consent was obtained from the patient.
Case report
A previously healthy 2-year-old female was admitted
to Tongji Hospital (Wuhan, China) with nausea and vomiting that had
lasted for five days. Upon physical examination, a bilateral
papilloedema was noted, but no other neurological deficits were
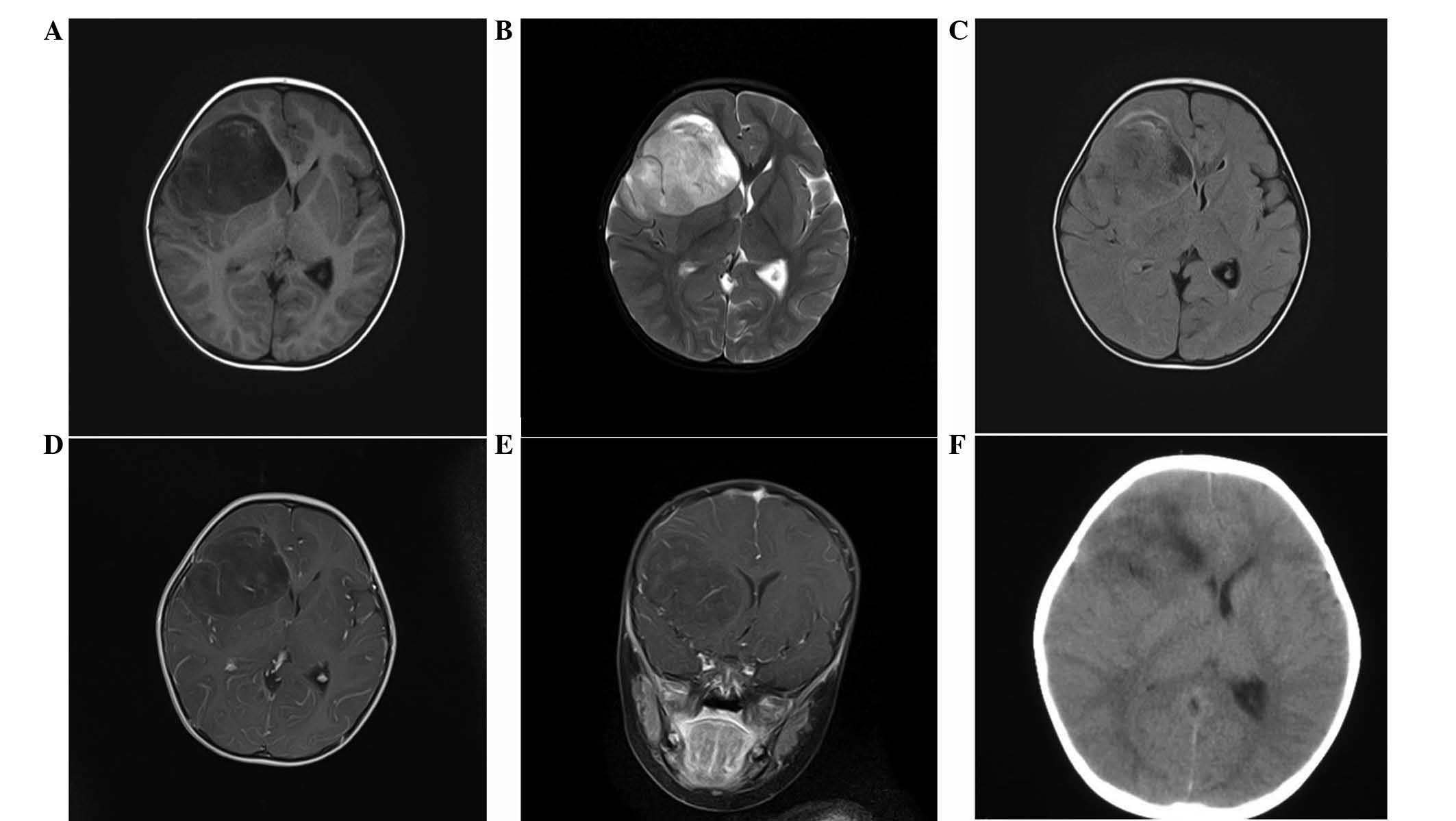
observed. A non-contrast head computed tomography (CT) scan showed
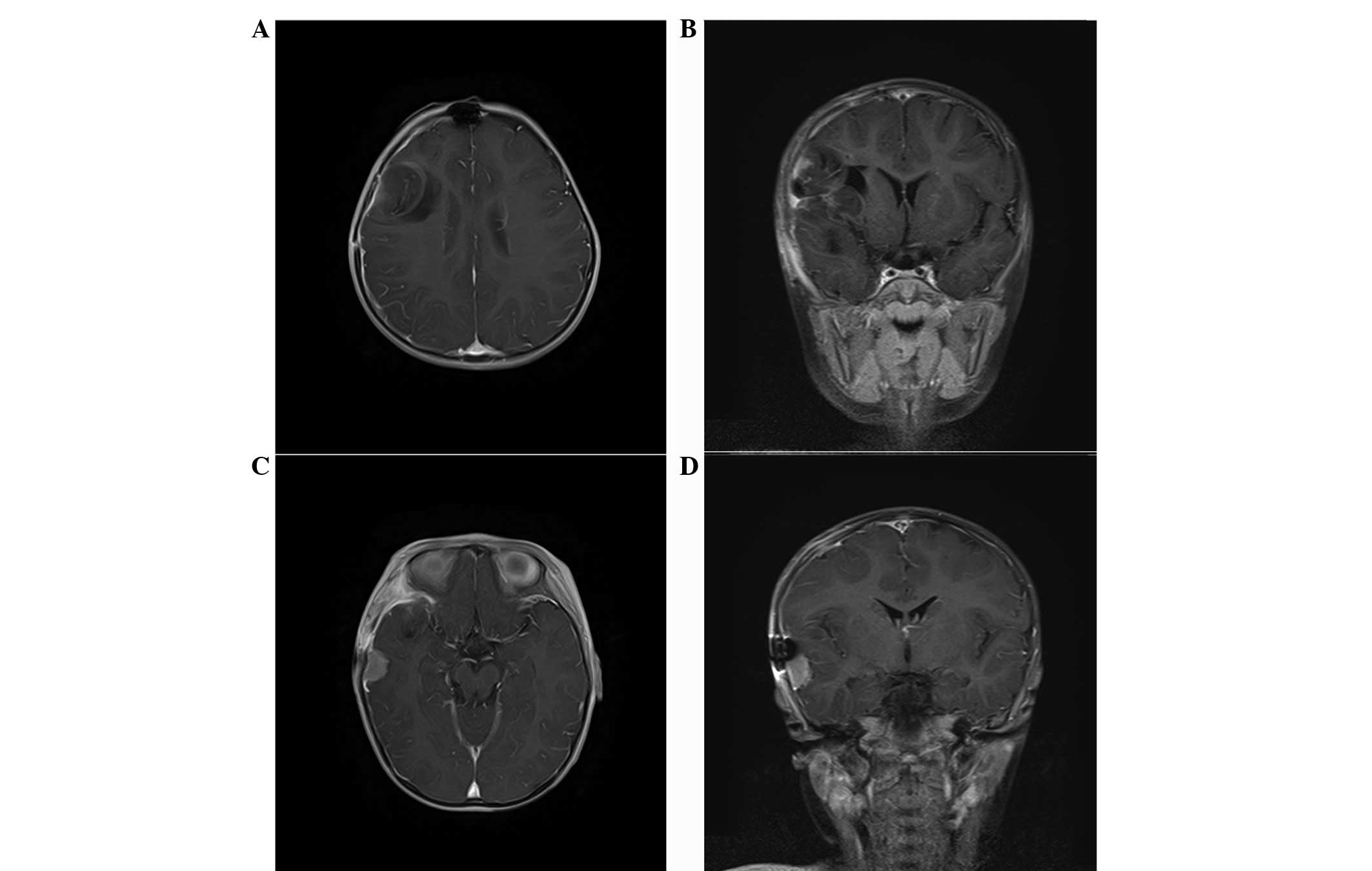
a large, microcystic and non-calcified right frontal lobe mass with
an ~10 mm right-to-left shift and effacement of the frontal horn of
the right lateral ventricle (Fig.
1). There was a small amount of vasogenic edema surrounding the
lesion, particularly on its medial aspect (Fig. 1).
Treatment with 20% mannitol (50 ml) and
dexamethasone (1 mh per 6 h) was initiated for the increased
intracranial pressure and cerebral edema. Magnetic resonance
imaging (MRI) revealed a T1 hypointense lesion with solid and
microcystic components and mild perilesional edema in the right
frontal lobe. Post-contrast, the lesion showed mild striped
enhancement, while restricted diffusion was observed in the solid
component (Fig. 1). There was a
mass effect on the ipsilateral lateral ventricle, a midline shift
of ~10 mm of the right frontal cortex and subfalcine herniation
(Fig. 1). On T2-weighted MRI, the
lesion was heterogeneously hyperintense and mild peritumoral edema
was identified in the frontal and temporal lobe (Fig. 1).
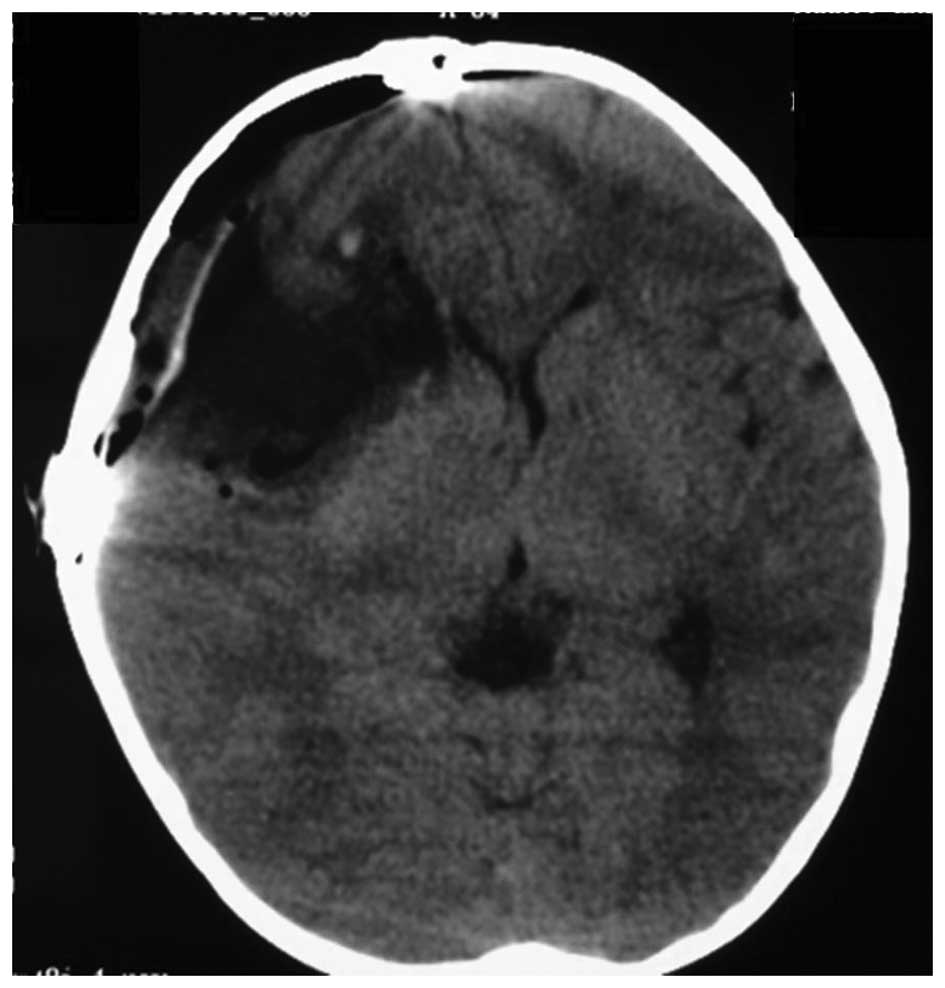
The patient underwent a right pterional craniotomy
for tumor resection. The tumor was not adherent to the overlying
parenchyma and there was not an excessive amount of vascularity.
The patient exhibited no neurological deficits post-operatively,
and post-operative CT showed gross total resection (GTR) of the
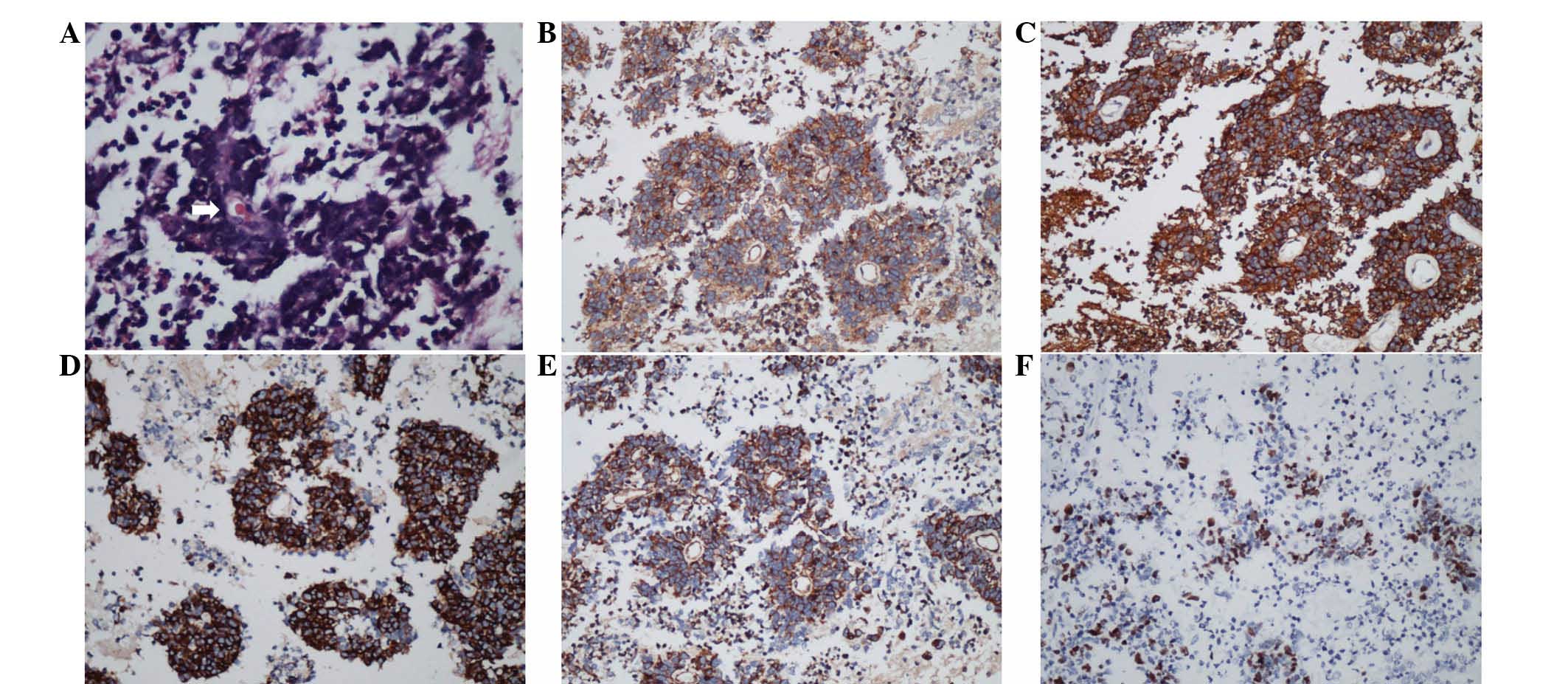
lesion (Fig. 2). Histologically,
the tumor exhibited uniform small round cells with regular nuclear
morphology (Fig. 3) and areas of
tumor apoplexy. Immunohistochemically, the tumor cells showed
perinuclear positivity for synaptophysin (Syn) and focal positivity
for oligodendrocyte transcription factor 2 (Olig2). In addition,
strong immunopositivity for nestin, microtubule-associated protein
2, vimentin and CD99 was noted. There was no positive staining for
glial fibrillary acidic protein (GFAP), epithelial membrane
antigen, neurofilaments and NeuN. The MIB-1 (Ki-67) labeling index
(LI) was 20% (Fig. 3). The
concluding histological diagnosis was atypical EVN (WHO grade
III).
Following surgery, adjuvant radiotherapy was refused
by the parents due to the patient’s young age. The patient
recovered well, remained neurologically intact and was discharged
from hospital 8 days after the surgery. However, at the 4-week
follow-up, the patient had developed a new-onset of generalized
tonic-clonic seizure episodes. Treatment with levetiracetam (200
mg/day) was initiated for seizure prophylaxis. Complete spinal
imaging was performed using MRI, with no evidence of long distance
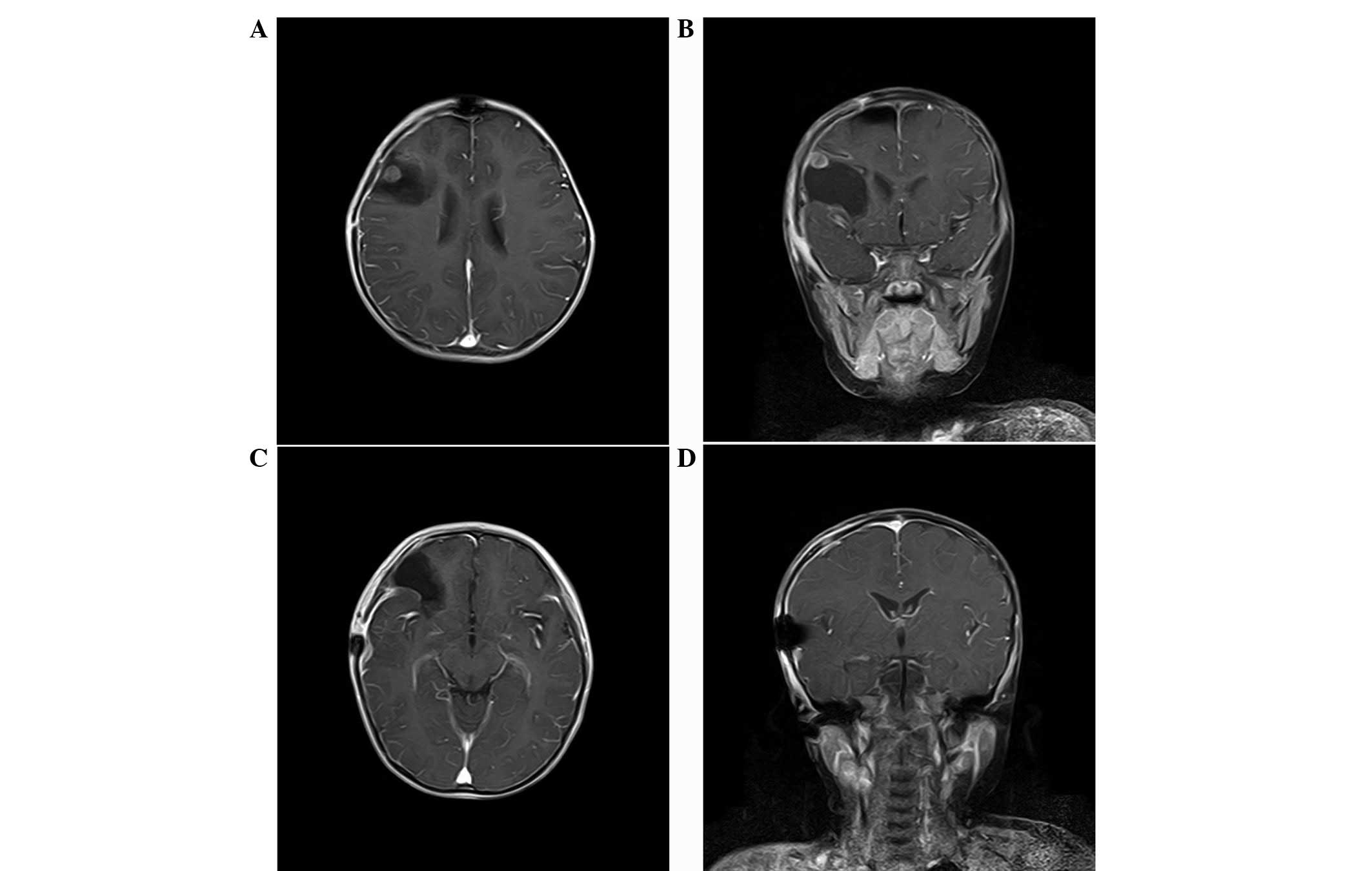
subarachnoid dissemination of the tumor being identified. However,
cerebral MRI revealed two small solid nodules along the
frontotemporal dura. One recurred in the initial operative area and
the other in the area posterior-inferior to the initial operative
area (Fig. 4). Adjuvant
radiotherapy and chemotherapy were refused by the patient’s parents
again. Ten weeks after the resection, the patient developed a
recurrence with nausea and vomiting, and MRI identified enlargement
of the two solid nodes (Fig. 5). A
craniotomy and tumor resection was refused. Following one week of
conservative therapy, the patient succumbed to EVN.
Discussion
To date, limited information on EVNs in the
pediatric population has been reported, and the literature is
composed almost exclusively of case reports. Tumors are classified
into typical or atypical categories, and atypical histological
criteria are designated by an MIB-1 LI of >3% or features
consistent with higher grade tumors (12). The current case report presents a
rare example of an atypical EVN in a 2-year-old female presenting
with new-onset nausea and vomiting. This case illustrates the fact
that atypical EVNs must be included as a possibility for a
non-calcified and microcystic parenchymal lesion in the pediatric
population. A literature review via the PubMed database was
performed using the keywords, ‘extraventricular’, ‘neurocytoma’,
‘children’ and ‘pediatrics’, alone and in combination.
Subsequently, the references in these studies were investigated for
additional reports. The search identified 28 studies reporting on
44 patients with EVNs that met the inclusion criteria (6–9,11,13–35).
In addition, three other reported pediatric cases were identified
in the Chinese literature (36–38).
For each study, the age, tumor location, treatment modality,
histopathological report, follow-up duration and recurrence data
were extracted for individual patients. Overall, 47 pediatric cases
were identified up to January, 2013. The tumor location is
summarized in Table I, and the
demographic data and clinical characteristics for patients with
atypical EVNs are summarized in Table
II.
 | Table ISummary of the frequency of EVNs
derived from 35 studies that met the inclusion criteria. |
Table I
Summary of the frequency of EVNs
derived from 35 studies that met the inclusion criteria.
| Reported
location | Lesions, n (%) |
|---|
| Frontal lobe | 16 (34) |
| Temporal lobe | 6 (13) |
| Parietal lobe | 5 (11) |
| Occipital lobe | 1 (2) |
| Frontoparietal
lobe | 2 (4) |
| Frontotemporal
lobe | 1 (2) |
| Parietotemporal
lobe | 1 (2) |
| Thalamus | 2 (4) |
| Cerebellum | 4 (9) |
| Vermis | 2 (4) |
| Spinal | 6 (13) |
| Mesencephalon | 1 (2) |
| Total | 47 (100) |
| Table IISummary of demographic and
lesion-associated data in cases of atypical EVNs reported in the
previous literature. |
Table II
Summary of demographic and
lesion-associated data in cases of atypical EVNs reported in the
previous literature.
| First author
(ref.) | Age,
years/gender | Tumor location | Resection | Calcification | Pathology | MIB-1, % | Adjuvant
therapy | Recurrence
time | Follow-up,
months/outcome |
|---|
| Ghosal et
al(20) | 9/M | Frontoparietal | ND | Yes | Syn (f), GFAP (f)
and NF (++) | 10.0 | ND | ND | ND |
| Singh et
al(31) | 8/M | Spinal D2-8 | STR | No | Syn (+), NSE (+),
GFAP (−), EMA(−) and NF (−) | 13.0 | No | ND | 3/normal |
| Raja et
al(30) | 7/M | Occipital | GTR | Yes | Syn (+), GFAP (±)
and NeuN (±) | 4.0 | Chemotherapy | 3 months | 6/cerebral spinal
dissemination |
| Mpairamidis et
al(25) | 3/F |
Parietotemporal | GTR | No | Syn (+), NeuN (+),
NF(−), β-tubulin III (−), MBP (−) and GFAP (±) | 5.0 | RT | ND | 12/normal |
| Choi et
al(18) | 8/M | Frontal | STR | Yes | Syn (++) | ND | γ-knife | 15 years | 12/second surgery
normal |
| Myung et
al(7) | 9/M | Frontal | STR | Yes | Syn (+), GFAP (+),
Olig2 (±), IDH1(−), p53 (−) and NeuN (−) | 16.8 | ND | 13.9 and 17.2
years | ND |
| Agarwal et
al(13) | 16/M | Spinal | STR | ND | Syn (+), GFAP (−),
NeuN (+) and NF (−) | 22.0 | RT | ND | 6/normal |
| Buchbinder et
al(16) | 1/F | Frontal | GTR | No | Syn (+) and GFAP
(−) | 10.0 | Chemotherapy | 2 months | 46/cerebral spinal
dissemination |
The collective case studies on EVNs indicated a
slight male predominance (29 males/18 females). Seizure activity
was the most common presenting symptom (15/47 patients), followed
by headaches (11/47 patients). These tumors may exhibit atypical
features consistent with aggressive clinical behavior. The tumors
were often cystic (16/47 patients) with frequent calcifications and
mild peritumoral edema. Garber and Brockmeyer reported that this
may aid the radiographical identification of EVNs from
oligodendrogliomas (19). By
contrast, Yi et al reported that the majority of the cases
(90%) demonstrated no peritumoral edema (35). No significant peritumoral edema was
identified in the patient of the present case report. Consistent
with the results of the present case, the majority of reported EVNs
are hypointense on T1-weighted MRI and hyperintense on T2-weighted
MRI, with heterogeneous enhancement following the administration of
a contrast agent (35). Therefore,
EVN clinically presents as a diagnostic challenge. The primary
differential diagnosis of EVN is oligodendroglioma with neurocytic
differentiation, oligoastrocytoma with neurocytic differentiation,
ganglioglioma, DNT and pineal parenchymal tumor of intermediate
differentiation (7).
In 1997, Giangaspero et al reported a series
of 11 patients diagnosed with EVNs (11). Three of the patients were children
with typical EVNs. Although one patient succumbed to EVN following
subtotal resection (STR) of a tumor located in the hypothalamus,
two underwent GTR with no evidence of disease after 29 and 31
months, respectively. Of the total 85 EVNs previously reported in
the literature, 27% were atypical (23/85 patients) (39). Atypical tumors exhibit 2–3 times the
recurrence risk of typical EVNs, as well as recurring at a much
earlier time post-treatment. A Kaplan-Meier analysis demonstrated
significant differences between patients with typical and atypical
tumors and post-primary treatment 5-year recurrence rates (36 and
68%, respectively; P<0.001) and 5-year mortality rates (4 and
44%, respectively; P<0.001) (39). However, complete resection may not
be possible due to the eloquence of surrounding structures or the
invasion of the surrounding periventricular parenchyma. This may
lead to a poorer prognosis in EVNs. In cases of typical EVNs,
overall recurrence rates have been recorded as 5% following GTR and
32% following STR (P<0.05; χ2 test). In cases of
incomplete resection, radiotherapy offers local control, but does
not appear to affect overall survival (11). STR with adjuvant radiotherapy was
associated with a 17% recurrence rate, which was not significantly
different when compared with STR alone. Disease progression was
observed in 20% of patients following subtotal resection at a mean
time of 30 months (40). As
observed in the present patient, adjuvant radiotherapy is advised
in cases following subtotal resection/biopsy. However, the value of
radiation therapy as adjunct therapy is debatable, as the majority
of clinicians agree that radiation therapy must be provided in
cases of incomplete resection or atypical histology (8,41). In
general, the literature on EVNs supports a total resection as an
optimistic treatment and the mandatory close follow-up of cases
with subtotal removal or aggressive histological features.
Post-operative chemotherapy and radiation are reserved for patients
that exhibit STR or recurrent disease.
EVN frequently demonstrates specific patently
neuronal features, including rosette and neuropil formation. This
is in addition to immune expression of multiple markers of neuronal
differentiation, particularly Syn in a diffuse manner (42). GFAP, Syn and NeuN are reliable
markers for glial and neuronal differentiation. Oligodendrogliomas
may possess neuronal differentiation and exhibit positive
immunostaining for markers of a neuronal tumor. Olig2 has been
identified as a transcription factor that regulates
oligodendroglial development and has been reported to be useful in
determining the diagnosis of oligodendroglioma (43,44).
In addition, central neurocytomas have been reported to be
immunohistochemically negative for Olig2 (44). Therefore, if tumor cells are
positive for Olig2, it is inappropriate to consider the tumor as a
neurocytoma. Despite immunostaining, it is difficult to
differentiate between oligodendrogliomas and EVNs. Studies
analyzing the genetic abnormalities of EVN have been seriously
limited. The codeletion of 1p19q has been described in a minority
of EVNs (45). Rodriguez et
al reported that the 1p19q codeletion occurred in 24% of EVNs
and that, as with oligodendroglial tumors, it was mediated in the
majority of cases by t(1;19)(42).
Commonly, the 1p19q codeletion predicts a more favorable prognosis
and an improved response to treatment in oligodendrogliomas,
however, this is not the case in EVNs (42). In the present literature review, the
codeletion was identified in 5/23 cases and all were adult
patients. Of the five patients, three exhibited recurrence and two
succumbed to EVN within 5 years of the initial tumor resection.
Four patients exhibited features of histological atypia by
pathological analysis, including necrosis and vascular hypertrophy.
This indicated that in EVNs, the presence of a 1p19q codeletion,
although rare, may imply aggressive clinical behavior and a poorer
outcome (46). Mutation of the
isocitrate dehydrogenase enzyme isoform 1 (IDH1 R132) and 2 (IDH2
R172) is associated with a large proportion of diffuse astrocytic
and oligodendroglial tumors. A recurrent mutation affecting codon
132 of the IDH1 gene, located on chromosome 2q33, is used for
differentiating oligodendroglioma-like tumors from others (47). High numbers of IDH1 point mutations
at codon 132 were observed in oligodendrogliomas and
oligoastrocytomas. All observed neurocytomas (central, n=35;
extraventricular, n=4) were negative for mutated IDH1 protein. In
an additional series of seven EVNs, IDH1 immunostaining was
negative for all cases, and IDH1 R132 and IDH2 R172 direct
sequencing revealed wild types in all cases (7). It was identified that the absence of
IDH1 expression functions as a powerful diagnostic indicator for
EVN-mimicking gliomas. A recent study by Yi et al also
reported observations of a series of IDH1 mutation-negative EVNs,
as determined by MRI (35).
Therefore, it may be hypothesized that the absence of the gene
mutation of IDH1 represents a prerequisite for EVN diagnosis. In
addition, the absence of p53 immunoexpression, MGMT promoter
methylation and a low frequency of EGFR gene amplification are
significant features of EVNs used to differentiate between
astrocytic and oligodendroglial tumors (7).
Neurocytomas are benign, slow-growing central
nervous system tumors of neuroglial origin (46). These tumors represent 0.25–0.5% of
all intracranial tumors in adults and an even smaller proportion of
pediatric CNS tumors. In 2001, Brat et al examined 33
pediatric and adult patients with EVNs (8). Within this study group, 14 patients
underwent GTR, whilst 19 patients underwent STR or biopsy. Of the
14 patients treated with GTR, no tumor was observed to recur during
the mean follow-up time of 29 months. Among the 19 patients who
underwent STR, three succumbed to EVN and ten exhibited recurrence,
with a median time to recurrence of 17 months. Patients with tumor
recurrence tended to exhibit a higher proportion of histological
atypia. Recurrence and mortality rates for typical CNs are not
dissimilar from those reported for typical EVNs, with 28%
recurrence and 5% mortality in typical CNs compared with 36%
recurrence and 4% mortality in typical EVNs (48). Atypical features appear to lead to
higher rates of recurrence and mortality in CNs and EVNs, with 40%
recurrence and 20% mortality in atypical CNs and 68% recurrence and
44% mortality in atypical EVNs (49). The extent of the resection appears
to be a significant prognostic indicator, as no completely resected
EVNs recurred, whereas >50% of patients who underwent STR
experienced recurrence. In addition, 12 cases of CN with
craniospinal dissemination following operative intervention have
been reported in the literature to date, indicating that an
aggressive, atypical form of this tumor may exist (50–52).
Stapleton et al recently reported a rare case in the
pediatric population of a diffuse CN with craniospinal
dissemination that was identified at the time of the initial
diagnosis by the immunohistochemical results of an elevated Ki-67
proliferation index (53). To date,
there have been five reported cases of EVN with craniospinal
dissemination (8,30,49,54–56),
including four males (7, 24, 48 and 75 years old) and one female
(71 years old). Two of the patients showed drop dural metastasis
and one tumor was initially located in the sellar region and was
associated with multiple remote disseminations in the spinal cord
and drop metastasis in the frontal cranial base in the route of the
initial surgery (54). The other
tumor was initially located in the left occipital-parietal lobe and
was associated with drop metastasis along the frontotemporal dura
with an intratumoral hemorrhage outside the field of the initial
surgery (56). The initial tumor of
the patient in the present case report was located in the right
frontal lobe. Four weeks after the initial surgery, cerebral MRI
revealed two small solid nodules along the frontotemporal dura, one
located in the field of surgery and the other located at the
trailing edge of the bone window. All tumors were considered
atypical with MIB-1 LI ranging between 4 and 30% (Table III). GTR was achieved in 4 of the
5 patients at the initial therapy, and additional radiation therapy
was delivered to one of the cases. However, the tumors recurred
several months later or disseminated into the craniospinal
cavities. A recent study by Kane et al observed that GTR and
STR with adjuvant radiotherapy appeared to offer improved
post-treatment tumor control rates for atypical EVNs (39). An additional recent study described
the use of GTR and STR with high dose chemotherapy, autologous stem
cell rescue and adjuvant therapy in a 9-month-old female with
recurrent atypical CN and leptomeningeal spread. The patient
exhibited a complete response to therapy and remained disease-free
at 4 years of age, until a recurrence 6 months later (16). These observations indicate that, for
atypical cases, long-term follow-up is required even when complete
remission has been achieved and novel treatment strategies,
including radiotherapy, chemotherapy and/or molecular targeted
therapies, have been used. The use of intensive chemotherapy
followed by autologous stem cell rescue for atypical neurocytoma
may be considered as an adjunct to surgical therapy in young
patients with atypical neurocytoma not amenable to radiation
therapy (16).
| Table IIIExtraventricular neurocytoma with
craniospinal dissemination. |
Table III
Extraventricular neurocytoma with
craniospinal dissemination.
| First author
(ref.) | Age,
years/gender | Tumor site | Initial
therapy | MIB-1, % | Follow-up,
months/outcome |
|---|
| Brat et
al(8) | 71/F | Cerebrum | Biopsy and RT | ND | 18/Succumbed to the
disease |
| Sharma et
al(55) | 24/M | Spine C5-T1 | GTR | 9 | 14/Cerebellar
metastasis |
| Raja et
al(30) | 7/M | Cerebrum | GTR | 4 | 6/Spinal canal
dissemination suspected |
| Wang et
al(56) | 75/M | Cerebrum | GTR and RT | >30 | 7/Dural
metastasis |
| Kawaji et
al(54) | 48/M | Sellar region | PR and RT | 3 | 6/Spinal canal
dissemination and metastasis in frontal cranial base |
| Present case | 3/F | Cerebrum | GTR | 20 | 1/Dural
metastasis |
EVNs are rare intraparenchymal lesions that must be
included in the differential diagnosis of a cerebral mass in
children. Although the imaging features of EVNs are variable, they
are usually cortically-based hemispheric lesions with variable
contrast enhancement and a cystic component, but they do not show
peritumoral edema. The frontal and temporal lobes are most commonly
involved. Immunohistochemically, EVNs are characterized by the
robust expression of Syn, but lack Olig2, IDH1 R132/IDH2 R172 and
p53 immunoexpression. The treatment for EVNs in the pediatric and
adult populations is GTR, with post-operative radiation reserved
for STR or recurrent disease. In addition, drop metastasis must be
carefully avoided. Recurrence and mortality rates remain high in
atypical EVN cases despite adjuvant radiation therapy. Therefore,
future studies must focus on determining successful chemotherapy
regimens and identifying novel molecular markers for targeted
adjuvant therapies.
Acknowledgements
The authors would like to thank Dr Pu Wang for
assisting with the writing and editing of the manuscript. The
current study was partially supported by a grant from the National
Natural Science Foundation of China (no. 81101620).
References
|
1
|
Hassoun J, Gambarelli D, Grisoli F, Pellet
W, Salamon G, Pellissier JF and Toga M: Central neurocytoma. An
electron-microscopic study of two cases. Acta Neuropathol.
56:151–156. 1982.PubMed/NCBI
|
|
2
|
von Deimling A, Janzer R, Kleihues P and
Wiestler OD: Patterns of differentiation in central neurocytoma. An
immunohistochemical study of eleven biopsies. Acta Neuropathol.
79:473–479. 1990.PubMed/NCBI
|
|
3
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, et al: The 2007 WHO classification of
tumours of the central nervous system. Acta Neuropathol.
114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McCutchen TQ, Smith MT, Jenrette JM, Van
Tassel P, Patel SJ and Thomas CR Jr: Interparenchymal hemorrhagic
neurocytoma: an atypical presentation of a rare CNS tumor. Med
Pediatr Oncol. 32:440–446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stephan CL, Kepes JJ, Arnold P, Green KD
and Chamberlin F: Neurocytoma of the cauda equina. Case report. J
Neurosurg. 90(2 Suppl): 247–251. 1999.PubMed/NCBI
|
|
6
|
Tortori-Donati P, Fondelli MP, Rossi A,
Cama A, Brisigotti M and Pellicanò G: Extraventricular neurocytoma
with ganglionic differentiation associated with complex partial
seizures. AJNR Am J Neuroradiol. 20:724–727. 1999.
|
|
7
|
Myung JK, Cho HJ, Park CK, Chung CK, Choi
SH, Kim SK and Park SH: Clinicopathological and genetic
characteristics of extraventricular neurocytomas. Neuropathology.
33:111–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brat DJ, Scheithauer BW, Eberhart CG and
Burger PC: Extraventricular neurocytomas: pathologic features and
clinical outcome. Am J Surg Pathol. 25:1252–1260. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Limaiem F, Bellil S, Chelly I, Mekni A,
Bellil K, Jemel H, et al: Extraventricular neurocytoma in a child
mimicking oligodendroglioma: a diagnostic pitfall. Pathologica.
101:105–107. 2009.PubMed/NCBI
|
|
10
|
Mut M, Güler-Tezel G, Lopes MB, Bilginer
B, Ziyal I and Ozcan OE: Challenging diagnosis: oligodendroglioma
versus extraventricular neurocytoma. Clin Neuropathol. 24:225–229.
2005.PubMed/NCBI
|
|
11
|
Giangaspero F, Cenacchi G, Losi L,
Cerasoli S, Bisceglia M and Burger PC: Extraventricular neoplasms
with neurocytoma features. A clinicopathological study of 11 cases.
Am J Surg Pathol. 21:206–212. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Söylemezoglu F, Scheithauer BW, Esteve J
and Kleihues P: Atypical central neurocytoma. J Neuropathol Exp
Neurol. 56:551–556. 1997.PubMed/NCBI
|
|
13
|
Agarwal S, Sharma MC, Sarkar C, Suri V,
Jain A, Sharma MS, et al: Extraventricular neurocytomas: a
morphological and histogenetic consideration. A study of six cases.
Pathology. 43:327–334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahmad F, Rosenblum MK, Chamyan G and
Sandberg DI: Infiltrative brainstem and cerebellar neurocytoma. J
Neurosurg Pediatr. 10:418–422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brandis A, Heyer R, Hori A and Walter GF:
Cerebellar neurocytoma in an infant: an important differential
diagnosis from cerebellar neuroblastoma and medulloblastoma?
Neuropediatrics. 28:235–238. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Buchbinder D, Danielpour M, Yong WH,
Salamon N and Lasky J: Treatment of atypical central neurocytoma in
a child with high dose chemotherapy and autologous stem cell
rescue. J Neurooncol. 97:429–437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheung YK: Central neurocytoma occurring
in the thalamus: CT and MRI findings. Australas Radiol. 40:182–184.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi H, Park SH, Kim DG and Paek SH:
Atypical extraventricular neurocytoma. J Korean Neurosurg Soc.
50:381–384. 2011. View Article : Google Scholar
|
|
19
|
Garber ST and Brockmeyer DL: A rare case
of a pediatric extraventricular neurocytoma: case report and review
of the literature. Childs Nerv Syst. 28:321–326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ghosal N, Dadlani R, Somorendra Singh S
and Hegde AS: Atypical extraventricular neurocytoma: a rare and
challenging case diagnosed on intraoperative cytology.
Cytopathology. 23:270–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hamilton R: Case of the month. August 1996
- frontal lobe tumor in 11 year old girl. Brain Pathol. 7:713–714.
1997.PubMed/NCBI
|
|
22
|
Harada M, Morioka T, Nishio S and Fukui M:
Neurocytoma in the left frontal lobe. No Shinkei Geka. 19:89–92.
1991.(In Japanese).
|
|
23
|
Makhdoomi R, Malik NK, Wani A, Bhat S and
Baba K: Extraventricular neurocytoma of the vermis in a child. J
Clin Neurosci. 17:1469–1471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Möller-Hartmann W, Krings T, Brunn A,
Korinth M and Thron A: Proton magnetic resonance spectroscopy of
neurocytoma outside the ventricular region - case report and review
of the literature. Neuroradiology. 44:230–234. 2002.PubMed/NCBI
|
|
25
|
Mpairamidis E, Alexiou GA, Stefanaki K,
Sfakianos G and Prodromou N: Extraventricular neurocytoma in a
child: case report and review of the literature. J Child Neurol.
24:491–494. 2009. View Article : Google Scholar
|
|
26
|
Nishio S, Takeshita I, Kaneko Y and Fukui
M: Cerebral neurocytoma. A new subset of benign neuronal tumors of
the cereb. Cancer. 70:529–537. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pal L, Santosh V, Gayathri N, Das S, Das
BS, Jayakumar PN and Shankar SK: Neurocytoma/rhabdomyoma
(myoneurocytoma) of the cerebellum. Acta Neuropathol. 95:318–323.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Polli FM, Salvati M, Miscusi M, Delfini R
and Giangaspero F: Neurocytoma of the spinal cord: report of three
cases and review of the literature. Acta Neurochir (Wien).
151:569–574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Psarros TG, Swift D, Mulne AF and Burns
DK: Neurocytoma-like neoplasm of the thoracic spine in a
15-month-old child presenting with diffuse leptomeningeal
dissemination and communicating hydrocephalus. Case report. J
Neurosurg. 103(2 Suppl): 184–190. 2005.
|
|
30
|
Raja AI, Yeaney GA, Jakacki RI, Hamilton
RL and Pollack IF: Extraventricular neurocytoma in
neurofibromatosis Type 1: case report. J Neurosurg Pediatr.
2:63–67. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Singh A, Chand K, Singh H, Sarkar C and
Sharma MC: Atypical neurocytoma of the spinal cord in a young
child. Childs Nerv Syst. 23:207–211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stapleton SR, David KM, Harkness WF and
Harding BN: Central neurocytoma of the cervical spinal cord. J
Neurol Neurosurg Psychiatry. 63:1191997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Treier M, Doostkam S and Meckel S:
Extraventricular neurocytoma: a rare cause of temporal lobe
epilepsy. Rofo. 183:1065–1066. 2011.(In German).
|
|
34
|
Yang GF, Wu SY, Zhang LJ, Lu GM, Tian W
and Shah K: Imaging findings of extraventricular neurocytoma:
report of 3 cases and review of the literature. AJNR Am J
Neuroradiol. 30:581–585. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yi KS, Sohn CH, Yun TJ, Choi SH, Kim JH,
Han MH, et al: MR imaging findings of extraventricular neurocytoma:
a series of ten patients confirmed by immunohistochemistry of IDH1
gene mutation. Acta Neurochir (Wien). 154:1973–1980. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Meng Z, Li M, Zhang Z and Liang S: MRI
diagnosis of extraventricular neurocytoma. Lin Chuang Fang She Xue
Za Zhi She. 4:441–444. 2010.(In Chinese).
|
|
37
|
Pan M, Fan Q, Zhang Z, Wang C, Zhou Z, Yu
M and Song G: Extraventricular neurocytoma of cerebellum in child:
a clinicopathologic analysis. Zhen Duan Bing Li Xue Za Zhi Bian Ji
Bu. 2:117–120. 2011.(In Chinese).
|
|
38
|
Yang Z and An Z: MRI scanning of one case
with extraventricular neurocytoma. Zhongguo Yi Xue Ying Xiang Xue
Za Zhi. 6:556–557. 2010.(In Chinese).
|
|
39
|
Kane AJ, Sughrue ME, Rutkowski MJ, Aranda
D, Mills SA, Lehil M, et al: Atypia predicting prognosis for
intracranial extraventricular neurocytomas. J Neurosurg.
116:349–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang YY, Kearney T, du Plessis D and
Gnanalingham KK: Extraventricular neurocytoma of the sellar region.
Br J Neurosurg. 26:420–422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sharma MC, Deb P, Sharma S and Sarkar C:
Neurocytoma: a comprehensive review. Neurosurg Rev. 29:270–285.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rodriguez FJ, Mota RA, Scheithauer BW,
Giannini C, Blair H, New KC, et al: Interphase cytogenetics for
1p19q and t(1;19)(q10;p10) may distinguish prognostically relevant
subgroups in extraventricular neurocytoma. Brain Pathol.
19:623–629. 2009. View Article : Google Scholar
|
|
43
|
Marie Y, Sanson M, Mokhtari K, Leuraud P,
Kujas M, Delattre JY, et al: OLIG2 as a specific marker of
oligodendroglial tumour cells. Lancet. 358:298–300. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yokoo H, Nobusawa S, Takebayashi H,
Ikenaka K, Isoda K, Kamiya M, et al: Anti-human Olig2 antibody as a
useful immunohistochemical marker of normal oligodendrocytes and
gliomas. Am J Pathol. 164:1717–1725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mrak RE, Yasargil MG, Mohapatra G, Earel J
Jr and Louis DN: Atypical extraventricular neurocytoma with
oligodendroglioma-like spread and an unusual pattern of chromosome
1p and 19q loss. Hum Pathol. 35:1156–1159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hallock A, Hamilton B, Ang LC, Tay KY,
Meygesi JF, Fisher BJ, et al: Neurocytomas: long-term experience of
a single institution. Neuro Oncol. 13:943–949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Capper D, Sahm F, Hartmann C, Meyermann R,
von Deimling A and Schittenhelm J: Application of mutant IDH1
antibody to differentiate diffuse glioma from nonneoplastic central
nervous system lesions and therapy-induced changes. Am J Surg
Pathol. 34:1199–1204. 2010. View Article : Google Scholar
|
|
48
|
Rades D, Fehlauer F, Lamszus K, Schild SE,
Hagel C, Westphal M and Alberti W: Well-differentiated neurocytoma:
what is the best available treatment? Neuro Oncol. 7:77–83. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rades D, Fehlauer F and Schild SE:
Treatment of atypical neurocytomas. Cancer. 100:814–817. 2004.
View Article : Google Scholar
|
|
50
|
Eng DY, DeMonte F, Ginsberg L, Fuller GN
and Jaeckle K: Craniospinal dissemination of central neurocytoma.
Report of two cases. J Neurosurg. 86:547–552. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ogawa Y, Sugawara T, Seki H and Sakuma T:
Central neurocytomas with MIB-1 labeling index over 10% showing
rapid tumor growth and dissemination. J Neurooncol. 79:211–216.
2006.PubMed/NCBI
|
|
52
|
Tomura N, Hirano H, Watanabe O, Watarai J,
Itoh Y, Mineura K and Kowada M: Central neurocytoma with clinically
malignant behavior. AJNR Am J Neuroradiol. 18:1175–1178.
1997.PubMed/NCBI
|
|
53
|
Stapleton CJ, Walcott BP, Kahle KT, Codd
PJ, Nahed BV, Chen L, et al: Diffuse central neurocytoma with
craniospinal dissemination. J Clin Neurosci. 19:163–166. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kawaji H, Saito O, Amano S, Kasahara M,
Baba S and Namba H: Extraventricular neurocytoma of the sellar
region with spinal dissemination. Brain Tumor Pathol. Dec
19–2012.Epub ahead of print.
|
|
55
|
Sharma S, Sarkar C, Gaikwad S, Suri A and
Sharma MC: Primary neurocytoma of the spinal cord: a case report
and review of literature. J Neurooncol. 74:47–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang L, Chen G, Wei H, Liu F, Hu H and
Zhang J: Dural metastasis of atypical extraventricular neurocytoma
with the codeletion of chromosomes 1p/19q. J Int Med Res.
39:2020–2026. 2011. View Article : Google Scholar : PubMed/NCBI
|