Introduction
Infantile myofibromatosis (IM), the most common
fibrous tumor of infancy, is a mesenchymal disorder that is
characterized by the proliferation of fibrous tumors in the skin,
bone, muscle and viscera (1). The
condition was first described by Stout in 1954 and was initially
termed ‘congenital generalized fibromatosis’ (2). The tumor was renamed by Chung and
Enzinger in 1981 to reflect its myofibroblastic characteristics
(3). The World Health Organization
(WHO), in the 2002 classification of soft tissue tumors, recognized
myofibromatosis under the benign category of
fibroblastic-myofibroblastic lesions (4). The condition usually presents prior to
the age of two years (3), but may
be observed in older children and even in adults (5). There are three distinct presentations,
solitary, multicentric without visceral involvement and
multicentric with visceral involvement. The solitary form tends to
occur predominately in males (6)
and is typically identified in the dermis, subcutis or deep soft
tissues. The reported incidence of solitary osseous myofibromatosis
is rare (7–9). The distribution is predominantly on
the head, neck and torso, with only a rare involvement of the
extremities (3). The present study
describes two cases of solitary IM involving the bones of the upper
extremities in females who were over two years old. The cases are
unusual in their symptom presentation and the origin of the tumor
is in a rarely observed location. Written informed consent was
obtained from the patients.
Case reports
Case 1
A three-year-old female patient was admitted to
Children’s Hospital of Zhejiang University School of Medicine
(Hangzhou, China) with a 10-day history of enlargement of a lump
located in the left forearm. The girl appeared systemically healthy
with a firm, well-circumscribed, subcutaneous nodule situated in
the left ulna and measuring 2×3 cm in dimension. The swelling was
slightly tender, but the overlying skin was not inflamed and normal
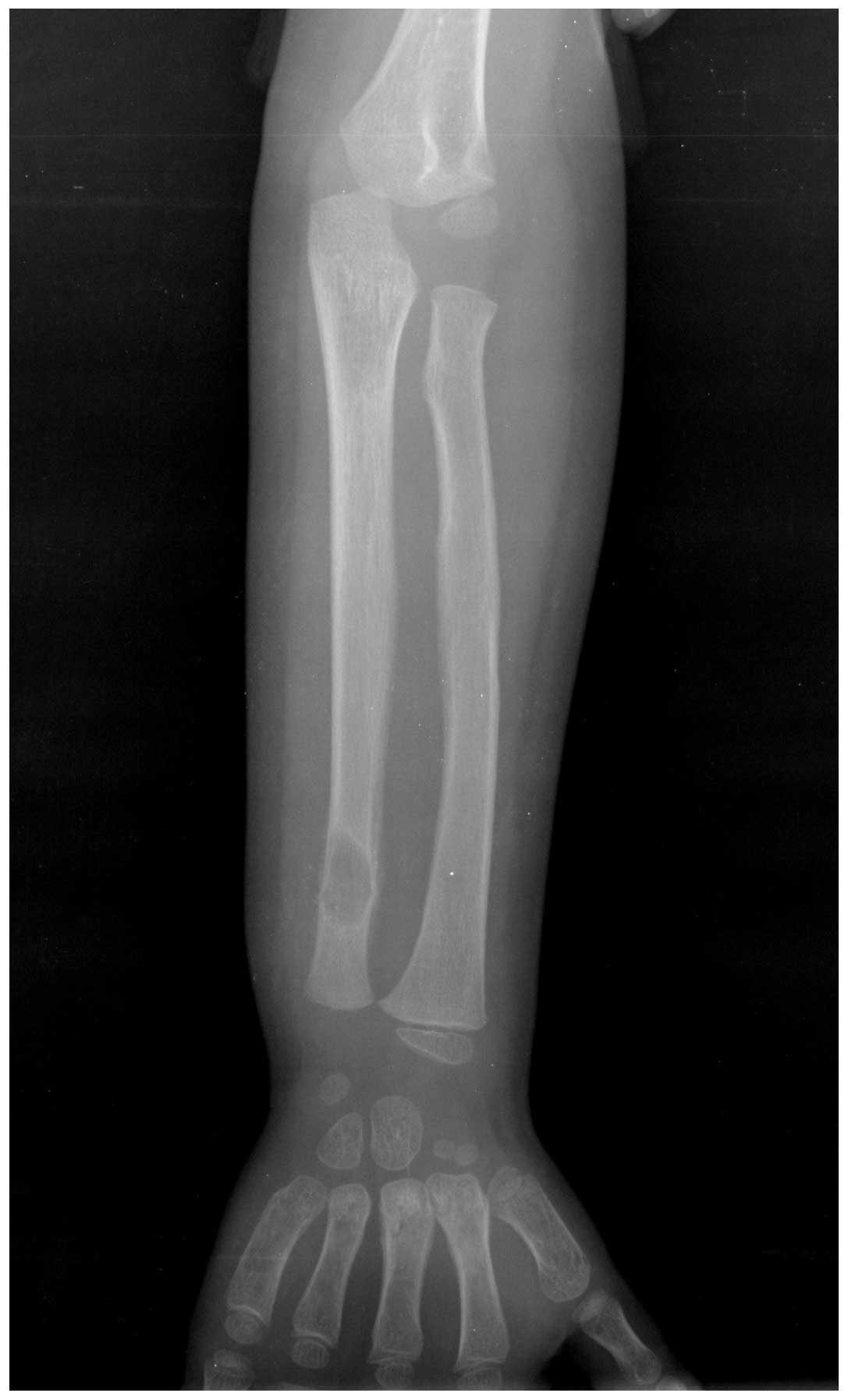
in color. The family history was unremarkable. An X-ray examination
exhibited a well-defined osteolytic lesion with slight marginal
sclerosis and a pathological fracture in the distal left ulna
(Fig. 1). To rule out further
tissue involvement, an ultrasound of the abdomen and a chest X-ray
were performed, which provided negative results. The patient
underwent curettage with bone grafting and flexible intramedullary
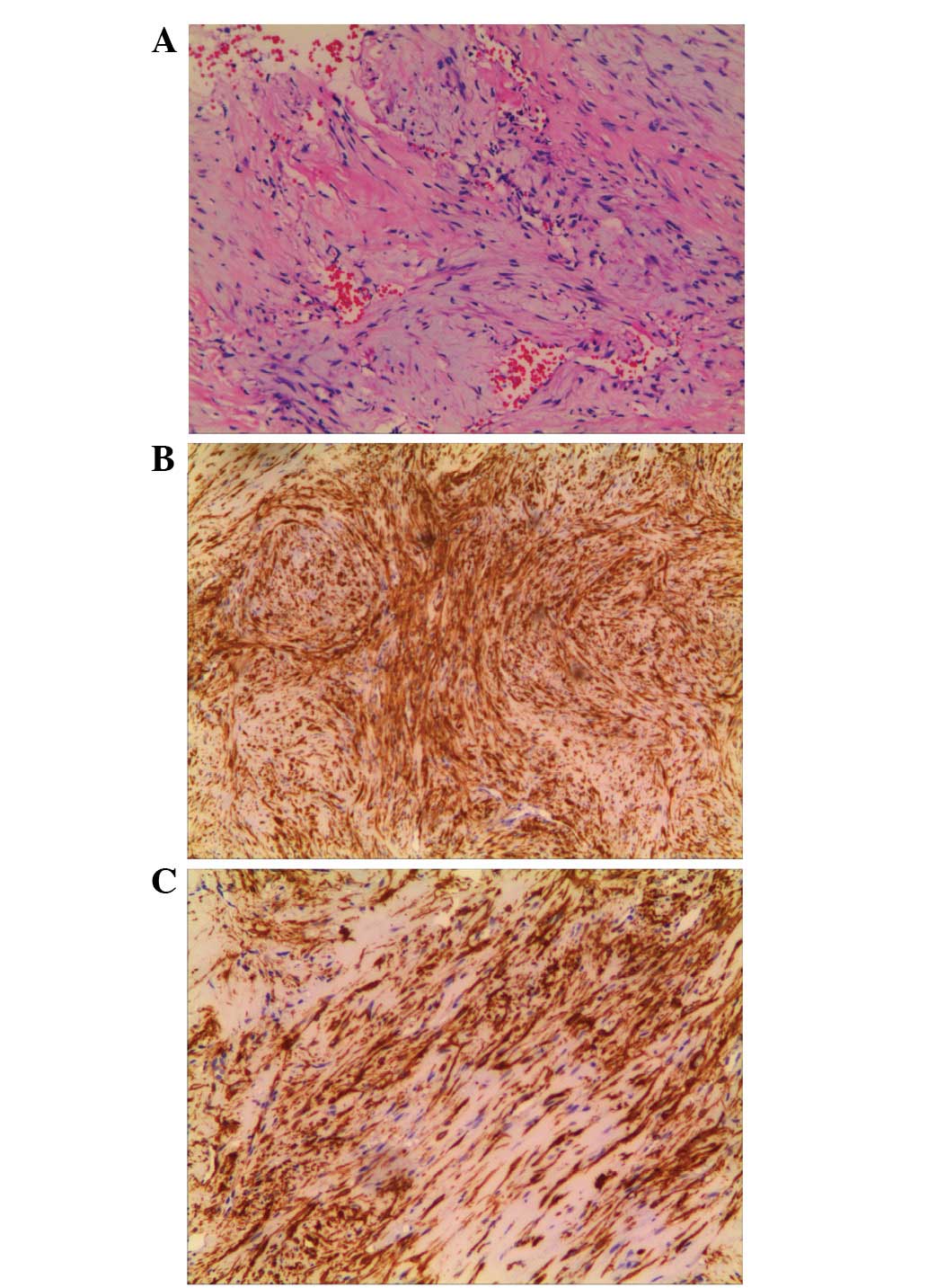
nail fixation on February 1, 2012. Histologically, the specimen
appeared to be formed of nodules that were composed of
cytologically bland spindle cells and abundant hyalinized stroma.
The cells were arranged in a fascicular and intertwining fashion
with minimal mitotic activities and without pleomorphism or atypia.
Blood vessels were abundant in a hemangiopericytoma-like pattern
(Fig. 2A). Immunohistochemistry
revealed positive staining for smooth muscle actin (SMA; Fig. 2B) and vimentin (VIM; Fig. 2C) and an absence of staining for
desmin and S-100. A diagnosis of myofibromatosis was formed. There
was no recurrence of any other lesions in the bone or soft tissues
during a one-year follow-up period.
Case 2
A nine-year-old female patient was sent to a local
clinic following a fall. The radiological examination revealed a
well-defined radiolucent lesion and a pathological fracture in the
diaphysis of the right humerus. Subsequent to being transferred to
Children’s Hospital of Zhejiang University School of Medicine for
further treatment, the patient was in a good general condition, but
was extremely vocal due to pain when the right arm was moved. The
family and antenatal histories were unremarkable. The blood tests
revealed a normal morphology and blood cell count. The laboratory
values for calcium and phosphate were normal. Additional imaging
studies failed to demonstrate any distant or visceral involvement.
The patient underwent curettage with bone grafting and flexible
intramedullary nail fixation on May 24, 2010. Macroscopically,
fascicles of spindle cells with abundant eosinophilic cytoplasm
resembled smooth muscle (Fig. 3).
The spindle and plump cells stained positively for SMA and VIM,
whereas desmin and S-100 markers were negative. The patient was
histopathologically diagnosed with solitary myofibromatosis. The
post-operative course was uneventful. During a two-year follow-up
period, no recurrence was identified either locally or
systemically.
Discussion
IM is a fibrous tumor of childhood and infancy that
is characterized by the development of nodular lesions involving
the skin, subcutaneous tissue, internal organs or bones. The tumor
may arise in a solitary or multicentric form, with similar
histopathological findings, but varied clinical features and
prognoses (5). Bone lesions are
seldom observed with the solitary type (5%), but are common with
the multicentric type (17–77%) (10–12).
The majority of the solitary IM cases of the bone have occurred in
the craniofacial bones (13). The
occurrence of solitary osseous myofibromatosis of the extremities
has been sporadically reported. When a review of the literature was
performed, one case of solitary IM in the appendicular bone of the
distal tibia was identified to have been reported by Inwards et
al(14), one case in the distal
ulna was reported by Kindblom and Angervall (15) and one case in the femur was reported
by Yamamoto et al(13). In
the two patients of the present study, the bone lesions of the
solitary type involved the ulna and the humerus, respectively.
The etiology of this disease is not well understood.
One of the proposed hypotheses is the upregulation of the estrogen
receptors on the fetal smooth muscles, which leaves them more
sensitive to maternal estrogen and may induce their proliferation
(16). Two other patterns of
inheritance have been described, autosomal dominant with low
penetrance and autosomal recessive (17). Studies with regard to the specific
genetic aberration have been limited, with monosomy 9q, trisomy 16q
and del(6)(q12;q15) being the few
cytogenetic abnormalities that have been reported (18,19).
The imaging characteristics of IM are not specific.
On radiographic images, the bone lesions appear as osteolytic areas
with sclerotic rims. On ultrasound scans, the masses may show
either a hyperechoic or anechoic center with a surrounding rim.
Computed tomography depicts the tumor as isodense or with a lower
density compared with the muscles, and bone involvement often takes
the shape of circumscribed lytic lesions with sclerotic margins
(20). Magnetic resonance imaging
of the tumor typically reveals a low intensity on T1-weighted
images and a high intensity on T2-weighted images.
Histologically, the cells have a characteristic
spindle shaped fibroblast appearance with pale pink cytoplasm and
elongated nuclei when stained with HE. Calcifications are frequent
observations and the mitotic activity is minimally increased
(21). The typical IM
immunohistological staining result is positive for VIM and SMA,
whereas it is negative for S-100 epithelial membrane antigen and
cytokeratin (22).
Myofibromas are frequently confused with a number of
other entities, including benign and malignant leiomyoma,
leiomyosarcoma, neurofibroma, fibrosarcoma, metastatic
neuroblastoma, hemangiopericytoma, desmoplastic fibroma and
inflammatory myofibroblastic tumors (23).
The prognosis of IM varies according to the type
(24). Tumors without visceral
involvement have an excellent outcome, with a spontaneous
regression of the lesions in one to two years. By contrast, IM with
visceral involvement is a severe disease. Gastrointestinal and
cardiopulmonary complications determine the early morbidity and
mortality of this IM presentation (24). Generally, the prognosis of solitary
IM of the bone is favorable. Inwards et al(14) reported no recurrences during the
post-operative follow-up of six cases. However, Kindblom and
Angervall (15) reported a case of
IM of the ulna that recurred twice following curettage. In the
present study, no recurrence was identified either locally or
systemically during the follow-up periods.
The treatment for IM is determined by the location
of the lesion. Although spontaneous regression is reported in a
large number of cases, recurrence has also been reported (10,11).
Surgical excision should be reserved for cases that affect the
vital functions (21). IM with
visceral involvement may require surgical or medical treatment,
including radiotherapy or chemotherapy with vincristine,
actinomycin D and cyclophosphamide, together with supportive care
(24). However, the literature is
unclear on the overall success of these alternative methods
(5). In the two patients of the
present study, the bone lesions had resulted in pathological
fractures and dysfunction of the forearms. The decision was
ultimately reached to treat with complete local excision with bone
grafting and flexible intramedullary nail fixation.
Although the incidence of solitary osseous
myofibromatosis is rare, IM should be considered in the
differential diagnosis for swellings and/or osteolytic lesions with
pathological fractures in a child’s extremities. Subsequent to
confirming a diagnosis, chest and abdominal imaging must be
performed to evaluate the overall prognosis and to direct treatment
(5). The patient should be followed
up to assess recurrences and to exclude the manifestation of
further nodules characterizing myofibromatosis (21).
References
|
1
|
Koujok K, Ruiz RE and Hernandez RJ:
Myofibromatosis: imaging characteristics. Pediatr Radiol.
35:374–380. 2005. View Article : Google Scholar
|
|
2
|
Stout AP: Juvenile fibromatoses. Cancer.
7:953–978. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chung EB and Enzinger FM: Infantile
myofibromatosis. Cancer. 48:1807–1818. 1981. View Article : Google Scholar
|
|
4
|
Murphey MD, Ruble CM, Tyszko SM,
Zbojniewicz AM, Potter BK and Miettinen M: From the archives of the
AFIP: musculoskeletal fibromatoses: radiologic-pathologic
correlation. Radiographics. 29:2143–2173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gatibelza ME, Vazquez BR, Bereni N, Denis
D, Bardot J and Degardin N: Isolated infantile myofibromatosis of
the upper eyelid: uncommon localization and long-term results after
surgical management. J Pediatr Surg. 47:1457–1459. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Josephson GD, Patel S, Duckworth L and
Goldstein J: Infantile myofibroma of the nasal cavity; a case
report and review of the literature. Int J Pediatr
Otorhinolaryngol. 74:1452–1454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sedghizadeh PP, Allen CM, Kalmar JR,
Miloro M and Suster S: Solitary central myofibroma presenting in
the gnathic region. Ann Diagn Pathol. 8:284–289. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shields CL, Husson M, Shields JA, Mercado
G and Eagle RC Jr: Solitary intraosseous infantile myofibroma of
the orbital roof. Arch Ophthalmol. 116:1528–1530. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tsuji M, Inagaki T, Kasai H, Yamanouchi Y,
Kawamoto K and Uemura Y: Solitary myofibromatosis of the skull: a
case report and review of literature. Childs Nerv Syst. 20:366–369.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wiswell TE, Davis J, Cunningham BE,
Solenberger R and Thomas PJ: Infantile myofibromatosis: the most
common fibrous tumor of infancy. J Pediatr Surg. 23:315–318. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wiswell TE, Sakas EL, Stephenson SR,
Lesica JJ and Reddoch SR: Infantile myofibromatosis. Pediatrics.
76:981–984. 1985.
|
|
12
|
Matthews MR and Cockerell CJ: An historic
perspective of infantile myofibromatosis. Adv Dermatol. 22:279–305.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamamoto T, Mizuno K and Hanioka K:
Solitary infantile myofibromatosis in the femur. Pathol Int.
50:255–257. 2000. View Article : Google Scholar
|
|
14
|
Inwards CY, Unni KK, Beabout JW and Shives
TC: Solitary congenital fibromatosis (infantile myofibromatosis) of
bone. Am J Surg Pathol. 15:935–941. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kindblom LG and Angervall L: Congenital
solitary fibromatosis of the skeleton: case report of a variant of
congenital generalized fibromatosis. Cancer. 41:636–640. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang W and Griffith K: Solitary
intestinal fibromatosis: a rare cause of intestinal obstruction in
neonate and infant. J Pediatr Surg. 26:1406–1408. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jennings TA, Duray PH, Collins FS, Sabetta
J and Enzinger FM: Infantile myofibromatosis: evidence for an
autosomal-dominant disorder. Am J Surg Pathol. 8:529–538. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stenman G, Nadal N, Persson S, Gunterberg
B and Angervall L: del(6)(q12q15) as the sole cytogenetic anomaly
in a case of solitary infantile myofibromatosis. Oncol Rep.
6:1101–1104. 1999.PubMed/NCBI
|
|
19
|
Sirvent N, Perrin C, Lacour JP, Maire G,
Attias R and Pedeutour F: Monosomy 9q and trisomy 16q in a case of
congenital solitary infantile myofibromatosis. Virchows Arch.
445:537–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Davies RS, Carty H and Pierro A: Infantile
myofibromatosis - a review. Br J Radiol. 67:619–623. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hausbrandt PA, Leithner A, Beham A, Bodo
K, Raith J and Windhager R: A rare case of infantile
myofibromatosis and review of literature. J Pediatr Orthop B.
19:122–126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beham A, Badve S, Suster S and Fletcher
CD: Solitary myofibroma in adult: clinicopathological analysis of a
series. Histopathology. 22:335–341. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Green MC, Dorfman HD, Villanueva-Siles E,
Gorlick RG, Thornhill BA, Weber RV and Geller DS: Aggressively
recurrent infantile myofibroma of the axilla and shoulder girdle.
Skeletal Radiol. 40:357–361. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Larralde M, Hoffner MV, Boggio P, Abad ME,
Luna PC and Correa N: Infantile myofibromatosis: report of nine
patients. Pediatr Dermatol. 27:29–33. 2010. View Article : Google Scholar
|