Introduction
Glioblastoma is the most common malignant tumor of
the central nervous system in adults. Glioblastoma is an invasive
tumor and should be regarded as a disease of the entire brain
(1,2). Despite recent advances in surgery,
radiotherapy and chemotherapy, there is no effective treatment for
glioblastoma, and the progression of the disease is almost always
the cause of mortality (3).
Radiotherapy is used in the treatment of this disease and shows
more potential as concomitant radio-chemotherapy. However,
radiotherapy with the highest doses is not possible beyond the
tolerance of normal brain tissue. Development of radiation
sensitizers that work on brain tumors is therefore required
(4,5). Histone deacetylase inhibitors (HDACIs)
induce growth arrest, differentiation and/or apoptosis of cancer
cells in vitro and in vivo(6–11).
Numerous HDACIs are under investigation in clinical trials, either
as monotherapies or in conjunction with other treatments, including
chemotherapy, biological therapy or radiation therapy (12). Valproic acid (VPA), a HDACI, is
commonly prescribed as an anti-epileptic drug in brain tumor
patients due to its effectiveness, oral bioavailability and
generally low toxicity profile (13–16).
VPA penetrates the blood brain barrier and is chronically
administered with minimal toxicity. As with other HDACIs, VPA
induces the cytotoxicity and apoptosis of tumor cells and
suppresses tumor invasion. Additionally, the action of VPA appears
to be specific to glioma cells (17).
Moreover, although VPA has been widely used for the
treatment of patients with gliomas, the cellular mechanisms
underlying the effects remain unknown. HDACIs enhance the radiation
response of various tumor cell types in vitro and in
vivo. The present study examined the effects of VPA and its
combination with radiation on the C6 glioma cancer cell line. The
effects on glioma cell viability and apoptosis were explored.
Materials and methods
Cell line and irradiation
The rat C6 glioma cell line (serial no.
3111C0001CCC000131) was obtained from the Cell Resource Centre
(Chinese Academy Of Medical Sciences, Beijing, China) and was
cultured in a humidified atmosphere of 95% air and 5%
CO2 (v/v) at 37°C in Dulbecco’s modified Eagle’s medium
[DMEM; 10% fetal bovine serum (FBS), 1% sodium-pyruvate, 1%
penicillin/streptomycin, 2 mmol/l L-glutamine, 0.1 mmol/l
non-essential amino acids and 1.5 g/l sodium bicarbonate; all from
HyClone, Logan, UT, USA]. Experiments were performed with cells
that were maintained in the exponential growth phase. VPA was added
prior to irradiation (3 Gy/min, 6 MV, X-ray), which was performed
in warm culture medium. VPA and MTT (Sigma, St. Louis, MO, USA)
were dissolved in phosphate-buffered saline (PBS) to a respective
stock concentration of 100 mmol/l and 50 mg/ml, then stored at
−20°C. The cells were irradiated using an Elekta Synergy
(Stockholm, Sweden) X-ray source. Dosimetry was performed by ion
chamber and chemical Fricke dosimetry.
MTT assay
To determine conditions for VPA treatment, specified
numbers (5×103) of cells in single cell suspensions were
seeded in individual wells of 96-well plates and incubated for 24 h
at 37°C prior to treatment with VPA at indicated concentrations
(0.25, 0.5, 1, 2 and 4 mmol/l) for specific time periods (24, 48
and 72 h). Following treatment, MTT solution was added to each well
and incubated for 4 h at 37°C prior to removal of the culture
medium. Dimethyl sulfoxide (DMSO) was then added and agitated for
30 min at room temperature. Cell viability was determined by
measuring the absorbance at 492 nm. Survival data were generated
after correcting for cell suppression from VPA alone. Cell
suppression ratio (%) = (1 − Ae/Ac) × 100, where Ae is the
absorbance of the experimental group and Ac is the absorbance of
the control.
Flow cytometry assay
Cells in the log phase were treated with VPA at
indicated concentrations (1×106/ml) or with PBS as a
control. Following incubation for an additional 48 h, cells at a
specific concentration (1×106/ml) were collected and
stained with Annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI) for 15 min at 4°C in the dark to identify
apoptotic cells by examining nuclear fragmentation and cell
membrane permeability under a fluorescence microscope, as per the
manufacturer’s instructions (MBL Co. Ltd., Woburn, MA, USA), at the
indicated time-points (4°C for 15 min). The extent of apoptosis was
quantified using a Becton-Dickinson (Franklin Lakes, NJ, USA) flow
cytometer and ModFit LT software (Verity Software House, Topsham,
ME, USA).
Clonogenic assay
To evaluate radiosensitivity, cells in the log phase
were plated into individual 25-ml cell culture flasks, which were
incubated for 24 h when the cells were attached, but not yet
divided. The cells were treated with VPA (0.5 mmol/l) or PBS as a
control for 24 h, followed by X-ray irradiation at various doses
(0, 2, 4, 6 and 8 Gy), and were then further incubated for 24 h in
the presence of the corresponding doses of VPA or not. Prior to
plating, the cell culture medium was removed and the cells were
washed twice with PBS. Adherent cells were then trypsinized and
counted, and five different numbers of cells per dose and substance
were seeded in triplicates into tissue culture dishes (Greiner
Bio-One, Frickenhausen, Germany) containing fresh, drug-free
culture medium without VPA. Colonies were allowed to form over 2–3
weeks. The cell culture medium was then removed and the cells were
washed twice with PBS. Colonies were fixed in 100% methanol for 30
min and stained with Giemsa for 15 min. The number of colonies
containing ≥50 cells was determined and the number of colonies was
normalized to that observed in the unirradiated controls. The
sensitizer enhancement ratio (SER) for VPA was calculated as the
ratio of the mean inactivation dose of the control divided by the
mean inactivation dose of VPA.
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Cells in the log phase were seeded into individual
cell culture flasks, which were incubated as aforementioned for 24
h. The cells were treated with VPA (0.5 mmol/l) or PBS as a control
for 24 h, followed by 4-Gy radiation or not, and were then
incubated for a further 24 h. Prior to isolation, the cell culture
medium was removed and the cells were washed twice with PBS. Total
RNA was extracted from the cells using TRIzol reagent (Takara
Holdings Inc., Otsu, Shiga, Japan) according to the manufacturer’s
instructions. cDNA was synthesized from 500 ng total RNA using
Takara reverse transcriptase and oligo(dT) primer in a total volume
of 10 μl. Reverse transcription was performed at 37°C for 50 min
followed by 15 min at 70°C for inactivation. PCR was performed with
aliquots of the cDNA samples at an annealing temperature of 60°C
with the following specific primers: Bcl-2 (228 bp) forward,
5′-CTGGTGGACAACATC GCTCTG-3′ and reverse, 5′-GGTCTGCTGACCTCACTT
GTG-3′; Bax (146 bp) forward, 5′-GCGAGTGTCTCCGGC GAATT-3′ and
reverse, 5′-GCCCCAGTTGAAGTTGCC ATCAG-3′; and GAPDH (146 bp)
forward, 5′-AAAAGGGTC ATCATCTCCG-3′ and reverse, 5′-AGTCTTCTGAGTGGC
AGTGAT-3′. Bax, Bcl-2 and GAPDH amplification reactions were
incubated for 15 min at 37°C and then for 30 sec at 95°C, followed
by 40 cycles of 95°C for 30 sec, 60°C for 34 sec and 72°C for 20
sec, and a final extension at 72°C for 10 min. Fold changes were
calculated in the following manner: Cycle number (Ct) of the target
genes was extrapolated from the software analysis program (SDS 1.9;
Applied Biosystems, Carlsbad, CA, USA) and then subtracted from the
Ct of the input control, and the difference in Ct was known as
Delta;Ct. All mean, standard error of the mean and statistical
values were calculated as a fold value. To determine the cell
radiosensitivity, controls without VPA or X-ray irradiation were
also included. Values for enrichment are expressed as a fold change
relative to the mean control value.
Immunoblot analysis
The status of Bax and Bcl-2 was determined by
immunoblot analysis. Cytosolic protein fractions and whole-cell
lysates for immunoblotting were prepared. For the whole-cell
lysates, the cells that were cultivated and treated as
aforementioned were lysed in RIPA buffer containing
phenylmethanesulfonyl fluoride after washing with cold PBS twice,
and then transferred to 1.5-ml microcentrifuge tubes. Five hours
after the delivery of irradiation, 1×107 cells were
harvested, washed with cold PBS and resuspended in 200 μl modified
KCl buffer. Samples were centrifuged at 21,000 × g for 20 min at
4°C. Equal amounts of proteins were separated by 15% SDS-PAGE and
transferred onto nitrocellulose membranes according to the
manufacturer’s instructions. The non-specific sites on the membrane
were blocked at 37°C for 2 h with 5% skimmed milk in Tris-buffered
saline supplemented with 0.1% Tween-20 (TBS-T) prior to being
washed twice in TBS-T. The membranes were incubated with either
rabbit polyclonal anti-Bax, anti-Bcl or goat anti-β-actin antibody
diluted in blocking solution overnight at 4°C. The membranes were
then washed three times in TBS-T and incubated with the
corresponding secondary antibody conjugated with horseradish
peroxidase at 1:2,000 dilution in blocking solution for 2.5 h at
room temperature. Subsequent to being washed thoroughly three times
in TBS-T, the membranes were developed by enhanced
chemiluminescence. β-actin was used as a loading control. Images
were processed with Image J software (National Institutes of
Health, Bethesda, MD, USA) for densitometric quantification. All
measurements were performed in triplicate.
Statistical analysis
For the statistical analyses, SAS software (version
9.1; SAS Institute, Cary, NC, USA) was used. All values are
expressed as the mean ± standard deviation. Mean values were
compared between three different groups using two-sample t-tests or
a one-way analysis of variance. P<0.05 was considered to
indicate a statistically significant difference.
Results
VPA reduces survival of C6 cells in a
dose- and time-dependent manner
VPA inhibited the proliferation of the rat C6 glioma
cells in a time- and dose-dependent manner, as determined by MTT
analysis (Table I). VPA at 0.25,
0.5, 1, 2 and 4 mmol/l decreased cell viability 24, 48 and 72 h
after spreading. The incubation of the cells with 0.25 mmol/l VPA
for 24 h resulted in only a modest decrease in cell survival.
However, 4 mmol/l VPA reduced cell survival to ~50% compared with
the untreated cells.
 | Table IVPA reduces survival of C6 cells in a
dose- and time-dependent manner. |
Table I
VPA reduces survival of C6 cells in a
dose- and time-dependent manner.
| VPA, mmol/l | Cell suppression
ratio, % |
|---|
|
|---|
| 24 h | 48 h | 72 h |
|---|
| 0.25 | 4.139±1.523a | 5.399±2.010a | 7.018±3.314a |
| 0.5 |
11.479±1.306a |
19.963±1.164a |
25.168±2.664a |
| 1 |
14.197±1.334a |
24.588±1.688a |
34.415±3.945a |
| 2 |
19.062±0.812a |
29.448±2.064a |
39.859±2.964a |
| 4 |
27.355±0.345a |
38.244±3.165a |
50.353±1.650a |
HDACIs are known to induce apoptosis in a variety of
cancer cell-lines (6,16). The present study further
investigated whether the decreased cell survival in the C6 cells
resulted from cell apoptosis. To further quantify cell apoptosis
induced by VPA, cell apoptosis was assayed by Annexin V-FITC
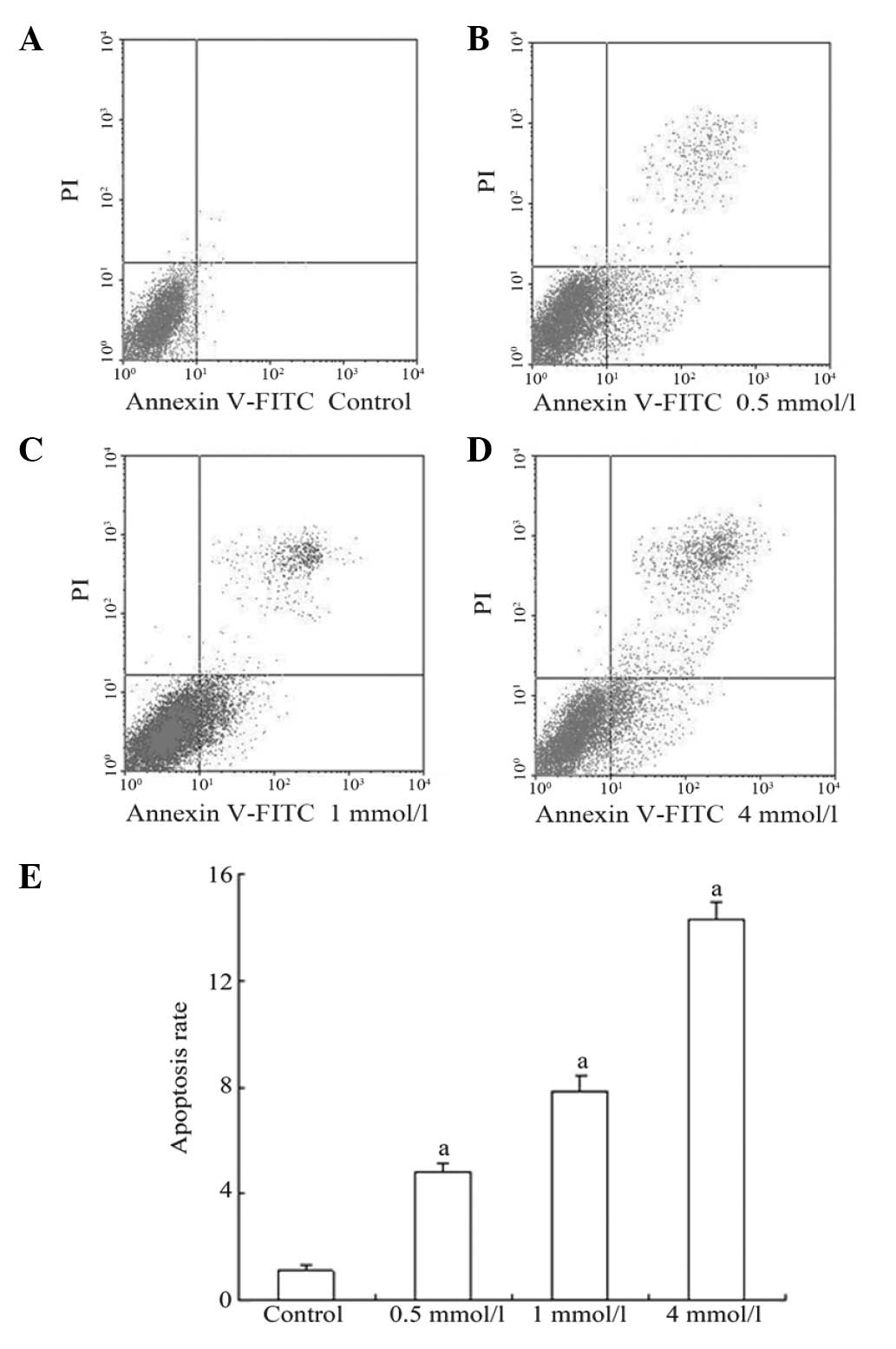
staining and flow cytometric analysis. As shown in Fig. 1, 0.5 mmol/l VPA treatment increased
the apoptotic rate from 1.133±0.166 to 4.768±0.348%. This rate was
further increased at 4 mmol/l.
Therefore, a final concentration of 0.5 mmol/l VPA
for 48 h was determined to be the optimal condition to treat the
cells.
VPA enhances the radiosensitization of C6
cells in a dose-dependent manner
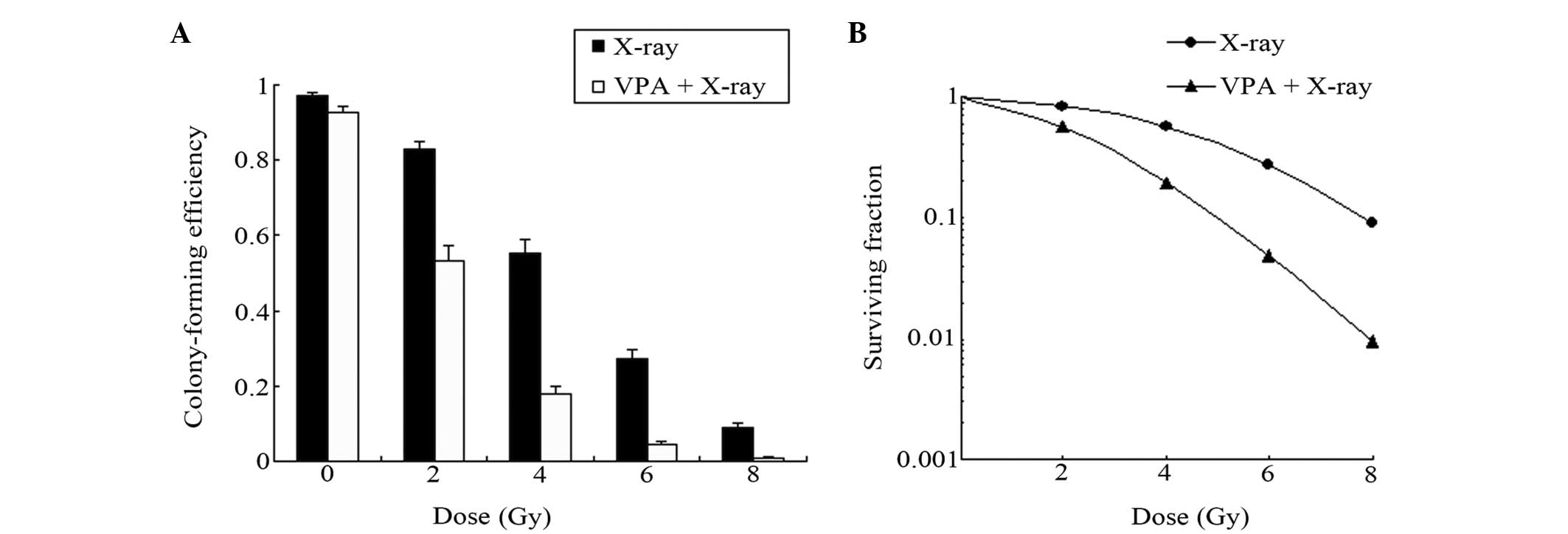
To determine whether VPA altered glioma cell colony
formation following irradiation in vitro, the effects of VPA
on tumor cell radiosensitivity were evaluated. Single cells were
seeded in culture conditions and irradiated with a single dose of
VPA (0.5 mmol/l) treatment for 48 h. This enhanced
radiation-induced cell death and the clonogenic formation at 4, 6
and 8 Gy radiation doses (Fig. 2).
VPA enhanced the radiosensitization of the C6 cells in a
dose-dependent manner at an SER. The cell SF curve was simulated by
a multitarget click mathematical model (Fig. 2), through which the related equation
and radioactivity parameters of are obtained. SF is obtained
according to the following equation: SF = colony forming numbers of
combined group/(numbers of inoculation cells × colony forming
numbers of cells without VPA) × 100. The cell survival curve is
obtained by taking the irradiation dosage as the abscissa axis and
SF as the vertical axis. Do and Dq may be calculated according to
the curve. Do represents the average lethal dosage of cells and Dq
represents the quasi-field dosage, which indicates the repair
ability of cells to sublethal injury. Subsequently the sensitizer
enhancement ratio (SER) may be calculated by the following
equation: SER = Do value of X-ray group/Do value of combined group
The SERs for combined X-ray and VPA treatment were 2.12 (Dq) and
1.84 (Do), which were 2.76 (Dq) and 2.39 (Do) in the X-ray group,
respectively. The SER for combined X-ray and VPA treatment,
compared to X-ray only, was 1.30.
VPA increases apoptotic responses to
X-ray irradiation by inhibiting Bcl-2 and increasing Bax
Next, the effects of VPA and X-ray exposure on the
mRNA and protein expression of substances known to be involved in
the apoptotic response were examined. The mRNA and protein levels
of Bax and Bcl-2 were determined by PCR and immunoblot
analysis.
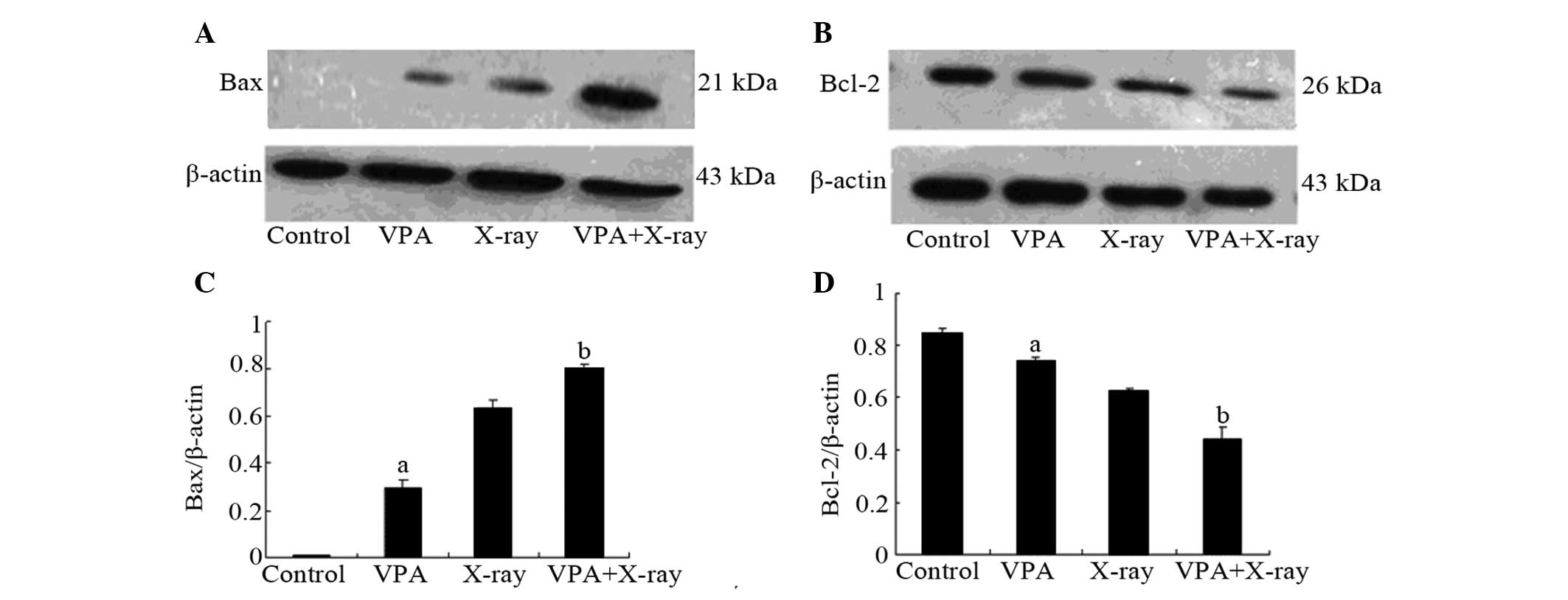
As shown in Table
II and Fig. 3, VPA and X-ray
exposure decreased the expression of Bcl-2. The combination of VPA
and X-ray further reduced the Bcl-2 mRNA and protein levels.
Accordingly, VPA and X-ray demonstrated increasing effects on Bax,
while the combination group further increased the Bax levels.
| Table IImRNA expression of Bcl-2 and Bax. |
Table II
mRNA expression of Bcl-2 and Bax.
| mRNA | Control | VPA | X-ray | VPA + X-ray |
|---|
| Bcl-2 | 0.816±0.051a | 0.652±0.055a | 0.508±0.046a | 0.247±0.041b |
| Bax | 0.236±0.045a | 0.339±0.025a | 0.528±0.049a | 0.856±0.034b |
Regardless of the level of transcription or the
level of translation, following treatment with VPA, the expression
of Bcl-2 mRNA and protein was decreased, whereas upregulation was
detected with Bax.
Discussion
Different HDACIs enhance the radiation response in a
variety of malignancies (18),
including human brain tumors (19–21),
head and neck squamous cell cancer (22), non-small cell lung cancer (23), colorectal cancer (24,25),
prostate cancer (9,20), melanoma (26) and metastatic breast cancer (27). However, the mechanisms contributing
to HDACI-induced radiosensitization have not been completely
identified. Defining the detailed mechanism would aid in the
clinical development of HDACIs as radiation sensitizers. The
present study further examined the radiosensitizing effects of VPA
on the C6 glioma cell line.
The results of this study demonstrated that exposure
to VPA leads to a decrease in clonogenic survival and an increase
in radiation-induced apoptosis in the rat C6 glioma cell line. VPA
also results in modulation of the radiation-induced mitochondrial
localization of Bcl-2 and Bax. Taken together, these data indicate
an important role of VPA-induced apoptosis in HDACI-mediated
radiosensitization.
In fact, histone modification not only enables
histone acetyltransferace and histone deacetylase disturbances to
lead to tumor development, but it is also able to alter the
acetylation status of numerous non-histone targets, including other
proteins involved in gene expression, DNA repair, proliferation,
migration, cell death, angiogenesis, inflammation and the immune
response. These targets are also likely to contribute to tumor
progression and resistance to treatments. Accordingly, HDACIs have
been shown, in a preclinical setting, to be effective anticancer
agents via multiple mechanisms, including the induction of cell
cycle arrest, intrinsic and extrinsic apoptotic mechanisms,
autophagic cell death, mitotic cell death, the generation of
reactive oxygen species, the inhibition of angiogenesis and
improvements in NK cell-mediated tumor immunity (6–11,28). A
study by Benitez et al(29)
demonstrated that VPA induces the neuronal-like differentiation of
astrocytoma cells. The MTT analysis in the present study showed
that VPA, one of the least potent classes of HDACIs (active only at
millimolar levels) (16), inhibited
the proliferation of rat C6 glioma cells in a time- and
dose-dependent manner, which started to become more evident at a
concentration of 0.5 mmol/l VPA for 48 h (Table I).
Radiotherapy remains one of the most common forms of
cancer treatment, leading to cell death through the induction of
DNA double-strand breaks (DSBs and the generation of reactive
oxygen species and reactive nitrogen species, which cause non-DNA
lesions or extracellular damage, such as lipid perioxidation
(30). DNA DSBs generated by
radiation may be the most lethal form of damage, although they may
be repaired via either homologous recombination or non-homologous
end-joining pathways (31,32). However, the lethality of radiation
is ascribed to unrepaired or insufficiently repaired DSBs. The
Dq-value of survival curves, or the so-called shoulder of survival
curves, represents at least in part the cells capability of
repairing DNA damage (Fig. 2),
although in certain studies there is evidence that cells with a
steep survival curve do not necessarily have a recovery deficiency
(33).
The exact mechanism of radiosensitization induced by
HDACIs was unknown until now. HDACIs were reported to prevent DSB
repair and prolong expression of γH2AX, a marker for DNA DSBs,
following radiation (34). A
variety of HDACIs have been shown to delay the dispersal of
radiation-induced γH2AX foci (19,20,23,26,35,36).
the present study analyzes the use of VPA in combination with
irradiation, focusing on the effects of HDACIs on DNA damage in
vitro. The SERs for combined treatment, compared with X-ray
only, confirm the above mechanism of radiosensitization to a
certain extent (Fig. 2). Thus, it
appears that VPA prevents DNA DSB repair, which leads to enhanced
glioma cell death. DNA damage then occurs as a consequence of the
HDACI-enhanced production of irradiation, even at low energy.
Previous studies have demonstrated that the
expression of Bax is increased following cell death stimulation,
and that Bax then translocates at the membrane of the mitochondria
to induce the release of cytochrome c(37,38).
Additionally, the cleaved form of Bax experiences
post-translational modification during apoptosis and is a potent
inducer of apoptosis (39,40). Bcl-2 has a role in anti-apoptotic
action by inhibiting the pro-apoptotic function of Bax. Cytochrome
c, which activates caspases, is released from the
mitochondria when the proportion of Bax/Bcl-2 increases. Caspases
are cysteine proteases that play a key role in cascade activation
during apoptosis induced by a number of stimuli (41–44).
To investigate the signaling pathways implicated in the
radiosensitization-induced apoptosis of the rat C6 cells, an
immunoblot analysis was used to examine the Bcl-2 and Bax protein
levels, while the mRNA levels were detected by qPCR. Bax expression
in the combined group was clearly upregulated relative to the other
groups, while Bcl-2 was apparently downregulated at the same time
(Table II and Fig. 3). Thus, radiosensitization-induced
apoptosis of rat C6 cells occurs, at least to a certain degree, via
the Bax/Bcl-2 pathway.
To summarize, VPA in combination with radiotherapy
exerts significant antitumor effects in vitro. Thus,
treatment with VPA in combination with radiotherapy may be a
notable approach for the inhibition of the growth of malignant
gliomas. Although promising as a sensitizer of radiotherapy,
further studies with VPA are warranted. An improved understanding
of the mechanisms of VPA-induced radiosensitization are likely to
help the development of fresh classes of drugs to treat human
diseases and exploit the characteristics of irradiation.
Additionally, it is important to investigate other more sensitive
HDACIs in combination with radiation to provide drugs that may be
better and more suitable for future clinical application.
Acknowledgements
This study was supported by Shandong Provincial
Natural Science Foundation, China (no. ZR2010HM080).
References
|
1
|
Agarwal S, Sane R, Oberoi R, Ohlfest JR
and Elmquist WF: Delivery of molecularly targeted therapy to
malignant glioma, a disease of the whole brain. Expert Rev Mol Med.
13:e172011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Berens ME and Giese A: ‘those left behind’
Biology and oncology of invasive glioma cells. Neoplasia.
1:208–219. 1999.
|
|
3
|
Régnard P, Bräuer-Krisch E, Troprès I,
Keyriläinen J, Bravin A and Le Duc G: Enhancement of survival of 9L
gliosarcoma bearing rats following intracerebral delivery of drugs
in combination with microbeam radiation therapy. Eur J Radiol.
68:S151–S155. 2008.PubMed/NCBI
|
|
4
|
Spence AM, Rasey JS, Dwyer-Hansen L, et
al: Toxicity, biodistribution and radioprotective capacity of
L-homocysteine thiolactone in CNS tissues and tumors in rodents:
comparison with prior results with phosphorothioates. Radiother
Oncol. 35:216–226. 1995. View Article : Google Scholar
|
|
5
|
Bernstein M, Cabantog AM, Glen J, Stiver S
and Mikulis D: Tirilazad does not protect rat brain from
brachytherapy-induced injury. Surg Neurol. 45:482–489. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marks P, Rifkind RA, Richon VM, Breslow R,
Miller T and Kelly WK: Histone deacetylases and cancer: causes and
therapies. Nat Rev Cancer. 1:194–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Johnstone RW: Histone-deacetylase
inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug
Discov. 1:287–299. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hess-Stumpp H: Histone deacetylase
inhibitors and cancer: from cell biology to the clinic. Eur J Cell
Biol. 84:109–121. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Camphausen K, Scott T, Sproull M and
Tofilon PJ: Enhancement of xenograft tumor radiosensitivity by the
histone deacetylase inhibitor MS-275 and correlation with histone
hyperacetylation. Clin Cancer Res. 10(18 Pt 1): 6066–6071. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheehan J, Ionescu A, Pouratian N, et al:
Use of trans sodium crocetinate for sensitizing glioblastoma
multiforme to radiation: laboratory investigation. J Neurosurg.
108:972–978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marks PA and Xu WS: Histone deacetylase
inhibitors: Potential in cancer therapy. J Cell Biochem.
107:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shabason JE, Tofilon PJ and Camphausen K:
Grand rounds at the National Institutes of Health: HDAC inhibitors
as radiation modifiers, from bench to clinic. J Cell Mol Med.
15:2735–2744. 2011. View Article : Google Scholar
|
|
13
|
Peterson GM and Naunton M: Valproate: a
simple chemical with so much to offer. J Clin Pharm Ther.
30:417–421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Phiel CJ, Zhang F, Huang EY, Guenther MG,
Lazar MA and Klein PS: Histone deacetylase is a direct target of
valproic acid, a potent anticonvulsant, mood stabilizer, and
teratogen. J Biol Chem. 276:36734–36741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Göttlicher M, Minucci S, Zhu P, et al:
Valproic acid defines a novel class of HDAC inhibitors inducing
differentiation of transformed cells. EMBO J. 20:6969–6978.
2001.PubMed/NCBI
|
|
16
|
Marks PA, Miller T and Richon VM: Histone
deacetylases. Curr Opin Pharmacol. 3:344–351. 2003. View Article : Google Scholar
|
|
17
|
Chen Y, Tsai YH and Tseng SH: Valproic
acid affected the survival and invasiveness of human glioma cells
through diverse mechanisms. J Neurooncol. 109:23–33. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Camphausen K and Tofilon PJ: Inhibition of
histone deacetylation: a strategy for tumor radiosensitization. J
Clin Oncol. 25:4051–4056. 2007. View Article : Google Scholar
|
|
19
|
Camphausen K, Cerna D, Scott T, et al:
Enhancement of in vitro and in vivo tumor cell radiosensitivity by
valproic acid. Int J Cancer. 114:380–386. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Camphausen K, Burgan W, Cerra M, et al:
Enhanced radiation-induced cell killing and prolongation of
gammaH2AX foci expression by the histone deacetylase inhibitor
MS-275. Cancer Res. 64:316–321. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JH, Shin JH and Kim IH: Susceptibility
and radiosensitization of human glioblastoma cells to trichostatin
A, a histone deacetylase inhibitor. Int J Radiat Oncol Biol Phys.
59:1174–1180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y and Jung M, Dritschilo A and Jung
M: Enhancement of radiation sensitivity of human squamous carcinoma
cells by histone deacetylase inhibitors. Radiat Res. 161:667–674.
2004. View
Article : Google Scholar
|
|
23
|
Geng L, Cuneo KC, Fu A, Tu T, Atadja PW
and Hallahan DE: Histone deacetylase (HDAC) inhibitor LBH589
increases duration of gamma-H2AX foci and confines HDAC4 to the
cytoplasm in irradiated non-small cell lung cancer. Cancer Res.
66:11298–11304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Folkvord S, Ree AH, Furre T, Halvorsen T
and Flatmark K: Radiosensitization by SAHA in experimental
colorectal carcinoma models - in vivo effects and relevance of
histone acetylation status. Int J Radiat Oncol Biol Phys.
74:546–552. 2009. View Article : Google Scholar
|
|
25
|
Chen X, Wong P, Radany E and Wong JY: HDAC
inhibitor, valproic acid, induces p53-dependent radiosensitization
of colon cancer cells. Cancer Biother Radiopharm. 24:689–699. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Munshi A, Kurland JF, Nishikawa T, et al:
Histone deacetylase inhibitors radiosensitize human melanoma cells
by suppressing DNA repair activity. Clin Cancer Res. 11:4912–4922.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baschnagel A, Russo A, Burgan WE, et al:
Vorinostat enhances the radiosensitivity of a breast cancer brain
metastatic cell line grown in vitro and as intracranial xenografts.
Mol Cancer Ther. 8:1589–1595. 2009. View Article : Google Scholar
|
|
28
|
López-Soto A, Folgueras AR, Seto E and
Gonzalez S: HDAC3 represses the expression of NKG2D ligands ULBPs
in epithelial tumour cells: potential implications for the
immunosurveillance of cancer. Oncogene. 28:2370–2382.
2009.PubMed/NCBI
|
|
29
|
Benitez JA, Arregui L, Cabrera G and
Segovia J: Valproic acid induces polarization, neuronal-like
differentiation of a subpopulation of C6 glioma cells and
selectively regulates transgene expression. Neuroscience.
156:911–920. 2008. View Article : Google Scholar
|
|
30
|
Yu H: Typical cell signaling response to
ionizing radiation: DNA damage and extranuclear damage. Chin J
Cancer Res. 24:83–89. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Santivasi WL and Xia F: The role and
clinical significance of DNA damage response and repair pathways in
primary brain tumors. Cell Biosci. 3:102013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Groselj B, Sharma NL, Hamdy FC, Kerr M and
Kiltie AE: Histone deacetylase inhibitors as radiosensitisers:
effects on DNA damage signalling and repair. Br J Cancer.
108:748–754. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cui YH, Liang HJ, Zhang QQ, et al:
Radiosensitivity enhancement by arsenic trioxide in conjunction
with hyperthermia in the EC-1 esophageal carcinoma cell line. Asian
Pac J Cancer Prev. 13:1693–1697. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Munshi A, Tanaka T, Hobbs ML, Tucker SL,
Richon VM and Meyn RE: Vorinostat, a histone deacetylase inhibitor,
enhances the response of human tumor cells to ionizing radiation
through prolongation of gamma-H2AX foci. Mol Cancer Ther.
5:1967–1974. 2006. View Article : Google Scholar
|
|
36
|
Zhang Y, Adachi M, Zhao X, Kawamura R and
Imai K: Histone deacetylase inhibitors FK228,
N-(2-aminophenyl)-4-[N-(pyri-
din-3-yl-methoxycarbonyl)amino-methyl]benzamide and
m-carboxycinnamic acid bis-hydroxamide augment radiation-induced
cell death in gastrointestinal adenocarcinoma cells. Int J Cancer.
110:301–308. 2004.PubMed/NCBI
|
|
37
|
Wolter KG, Hsu YT, Smith CL, Nechushtan A,
Xi XG and Youle RJ: Movement of Bax from the cytosol to
mitochondria during apoptosis. J Cell Biol. 139:1281–1292. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Putcha GV, Deshmukh M and Johnson EM Jr:
BAX translocation is a critical event in neuronal apoptosis:
regulation by neuroprotectants, BCL-2, and caspases. J Neurosci.
19:7476–7485. 1999.PubMed/NCBI
|
|
39
|
Thomas A, El Rouby S, Reed JC, et al:
Drug-induced apoptosis in B-cell chronic lymphocytic leukemia:
relationship between p53 gene mutation and bcl-2/bax proteins in
drug resistance. Oncogene. 12:1055–1062. 1996.PubMed/NCBI
|
|
40
|
Yanase N, Takada E, Yoshihama I, Ikegami H
and Mizuguchi J: Participation of Bax-alpha in IFN-alpha-mediated
apoptosis in Daudi B lymphoma cells. J Interferon Cytokine Res.
18:855–861. 1998. View Article : Google Scholar
|
|
41
|
Lin HI, Lee YJ, Chen BF, et al:
Involvement of Bcl-2 family, cytochrome c and caspase 3 in
induction of apoptosis by beauvericin in human non-small cell lung
cancer cells. Cancer Lett. 230:248–259. 2005.PubMed/NCBI
|
|
42
|
Kim DS, Jeon SE, Jeong YM, Kim SY, Kwon SB
and Park KC: Hydrogen peroxide is a mediator of indole-3-acetic
acid/horseradish peroxidase-induced apoptosis. FEBS Lett.
580:1439–1446. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zuliani T, Obriot H, Tual M, et al:
Variable Bax antigenicity is linked to keratinocyte position within
epidermal strata and UV-induced apoptosis. Exp Dermatol.
17:125–132. 2008. View Article : Google Scholar
|
|
44
|
Park SK, Kang H and Kwon CH:
Caspase-dependent cell death mediates potent cytotoxicity of
sulfide derivatives of 9-anilinoacridine. Anticancer Drugs.
19:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|