Introduction
Therapeutic agents that target tumor specific
antigens have the potential to produce effective treatment while
minimizing side-effects. Combining therapeutic agents that target
more than one tumor antigen may enhance the killing of tumor cells.
In addition, a combination of therapeutic agents, targeting
multiple antigens, may prevent the escape of tumor cells if one of
the antigens is no longer presented. One such tumor antigen that is
overexpressed on tumor cells is the epidermal growth factor
receptor (EGFR) (1). Targeting may
be via the ligand for EGFR, EGF. Fusion of a toxin to a ligand for
the receptor produces a specifically targeted cytotoxic agent
(1). Receptor-mediated endocytosis
internalizes the toxin, which causes cell death (2). One such toxin is Pseudomonas
exotoxin (PE) (1). Tumor growth
factor α (TGFα) is similar in structure to EGF and binds to EGFR at
a similar rate (3). Since
conjugation of PE to EGF is inefficient, TGFα is often selected as
an alternative (1). TGFα-PE38 is
cytotoxic to tumor cells in vitro (4). Furthermore, a preclinical study in
athymic, nude mice demonstrated the effectiveness of TGFα-PE38 in
reducing the size of human brain tumor cell lines (5). Based on these studies, TGFα-PE38 was
selected as one of the targeted therapies.
The second targeted therapy was that of mucin-1
(MUC1)-stimulated peripheral blood mononuclear cells (PBMCs;
M1SMC), which produce cytotoxic T lymphocytes (CTLs) that kill
human breast cancer cells in vitro (6) and prevent human breast cancer cell
tumor development in vivo in non-obese diabetic, severe
combined immunodeficient (NOD-SCID) mice (7).
Materials and methods
Human cells
All human cells were obtained from deceased subjects
in accordance with the Texas Tech University Health Sciences Center
institutional review board. The study was approved by the ethics
committee of Texas Tech University Health Sciences Centre
(Amarillo, TX, USA). Frozen human peripheral blood hematopoietic
stem cells (HSCs) were obtained from the Bone Marrow Transplant
Laboratory of the Harrington Cancer Center (Amarillo, TX, USA) from
deceased, anonymous donors. Frozen PBMCs were acquired by apheresis
from a deceased, anonymous donor with breast adenocarcinoma.
TGFα-PE38, a gift from Ira Pastan, National Cancer
Institute (Bethesda Maryland, USA), was aliquoted and stored at
−70°C.
MUC1 peptides
A single repeat of the MUC1-variable number tandem
repeat (VNTR)1 peptide, GSTAPPAHGVTSAPD TRPAP (8), was synthesized by American Peptide
Co., Inc. (Sunnyvale, CA, USA).
Cell culture conditions
Procedures were performed as described previously
(6). The PBMCs were not HLA typed,
since our previous studies (7,9,10) and
other studies (11) have found that
cytotoxicity by M1SMC may be non-major histocompatibility complex
(MHC)-restricted. Cells were cultured at 2×106 cells/ml
in AIM-V® serum-free lymphocyte medium (GIBCO-BRL, Life
Technologies Inc., Grand Island, NY, USA) and maintained in a 37°C
humidified 5% CO2 atmosphere (12). Interleukin (IL)-2 (Cetus
Corporation, Berkeley, CA, USA) was added twice per week at 100
IU/ml. The cells were stimulated with MUC1-VNTR1 peptide on days
zero and seven, at 1 μg/ml. The cells were harvested on day eight.
HSCs were cultured at 2×106 cells/ml in
AIM-V®.
Cytotoxicity assays
The MCF7 (HLA-A2) breast cancer cell line was
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA) and cultured as recommended. MCF7 cells express
hypoglycosylated mucin (13). This
cell line was used as the target cell line in an XTT®
assay (Roche Diagnostics Corp., Indianapolis, IN, USA) (14,15)
performed according to the manufacturer’s instructions. The
TGFα-PE38 or effector cells were tested at a concentration of
effector-to-target cell ratio of 1.25. Medium was added in place of
effector cells to the spontaneous release control wells. The
effector and target cells were incubated together for 18 h, which
we had previously found to be superior to 4 h (6), at 37°C and 5% CO2. This was
performed in triplicate. The percentage of specific lysis (%SL) =
[OD(target - medium) − OD(A – B)]/OD(target - medium) ×100, where A
represents the experimental (target plus effectors) wells and B
represents the wells with a corresponding number of effectors.
Other assays
Other assays included the Trypan Blue Cell Viability
Solution assay (Sigma-Aldrich, St. Louis, MO, USA) (16), the DePsiphler Mitochondrial
Potential assay (R&D Systems, Inc., Minneapolis, MN, USA)
(17) and the in situ
chromium uptake assay (18),
performed according to the manufacturer’s instructions.
Progenitor assay
The cells to be tested for progenitor cell content
were thawed and washed to remove cryoprotectant. The cells were
counted and added to cultures at 5×105 cells/ml in
methylcellulose (MethoCult media™; Stem Cell Technologies,
Vancouver, BC, Canada) according to the manufacturer’s instructions
(19). Cultures were examined after
14 days and scored for the presence of erythroid [burst forming
unit-erythroid (BFU-E)] and myeloid (colony-forming
unit-granulocytes, monocytes (CFU-GM)] progenitor colonies.
Statistical analysis
Fisher’s exact test, the χ2 test, the
Kruskal-Wallis test and the Mann-Whitney signed rank test were used
to analyze data. P<0.05 was considered to indicate a
statistically significant difference.
Results
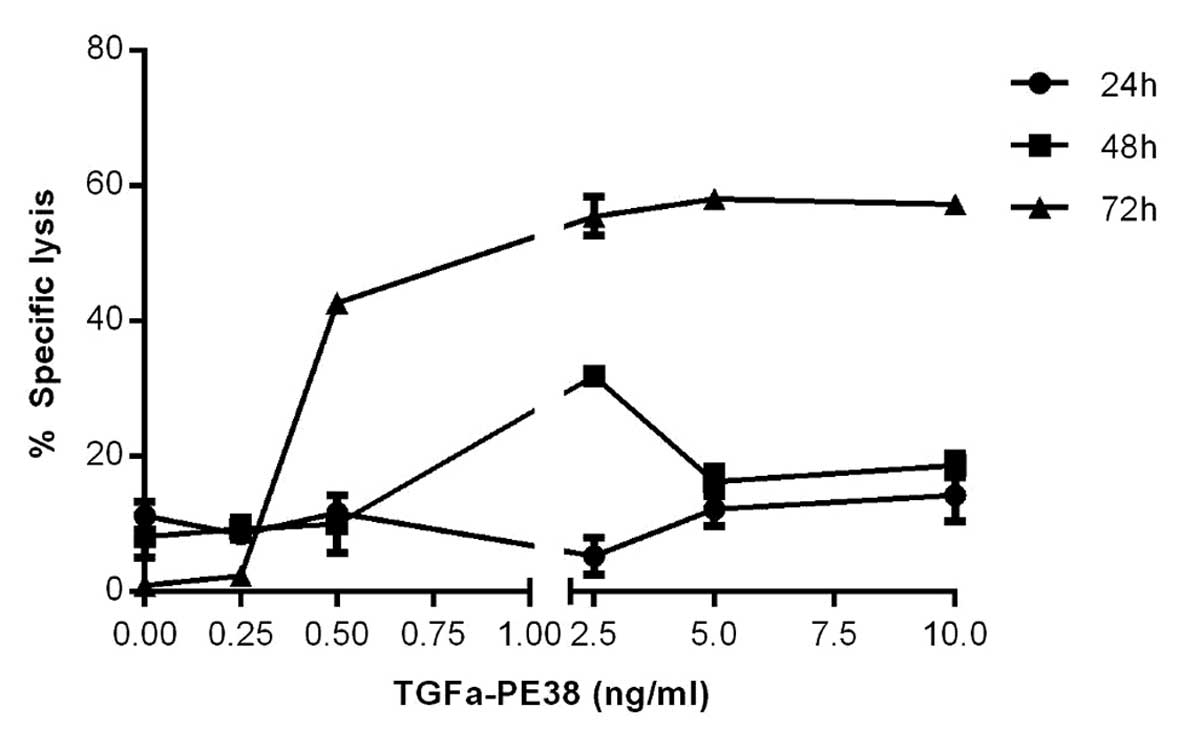
Concentration- and time-dependent
specific lysis of MCF7 cells by TGFα-PE38
Based upon the reported IC50
(concentration of immunotoxin causing a 50% reduction in protein
synthesis) for MCF7 cells of 1.1 ng/ml when using TGFα-PE38
(4), an analysis of the %SL of the
MCF7 cells at a range of TGFα-PE38 concentrations below and above
this value were performed to determine the optimum concentration
for use in combination with CTLs. The 50% lysis of the MCF7 cells
by TGFα-PE38, which was between 0.5 and 2.5 ng/ml at 72 h of
incubation (Fig. 1), agreed with
the reported IC50 data (4). Other assays, including the Trypan Blue
Cell Viability Solution assay (16), the DePsiphler Mitochondrial
Potential assay (17) and the in
situ chromium uptake assay (18), produced similar results, but with a
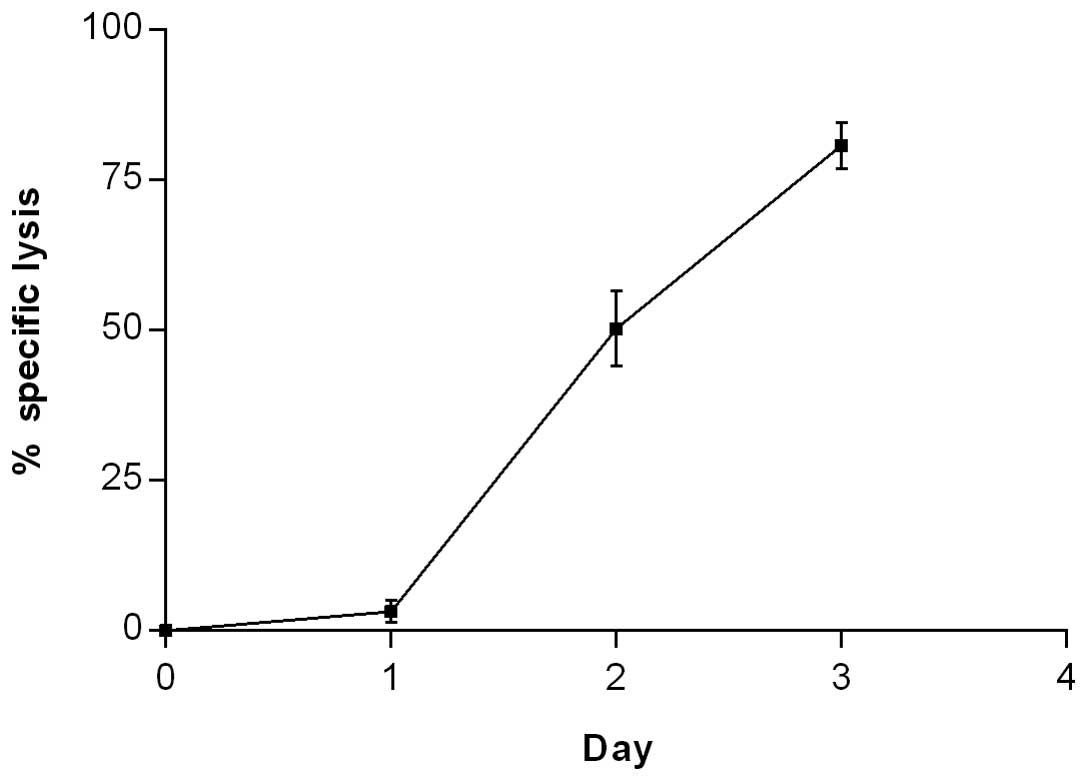
wider range of error. The %SL plateaued at 5 ng/ml TGFα-PE38
(Fig. 1) and thus was used for the
study of time-dependent specific lysis of the MCF7 cells by
TGFα-PE38. There was little (3%) specific lysis of the MCF7 cells
by TGFα-PE38 at 24 h, however, this increased exponentially to 50
and 81% at 48 and 72 h, respectively (Fig. 2).
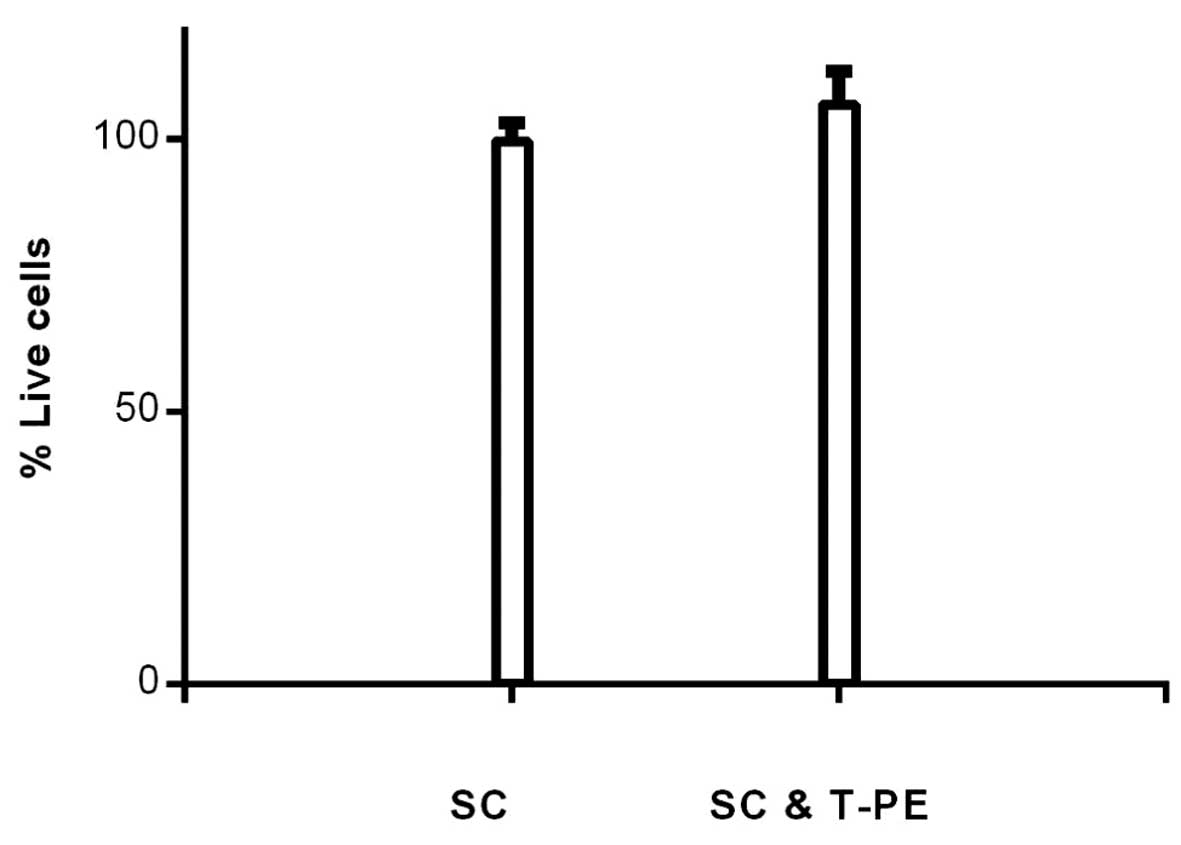
Effect of TGFα-PE38 on HSC viability
To determine if TGFα-PE38 was non-toxic in normal
cells, frozen human peripheral blood HSCs were thawed and incubated
with or without 5 ng/ml TGFα-PE38 for five days. The five-day
incubation time was selected, as we had shown that the majority of
MCF7 cells were killed by TGFα-PE38 at 72 h. The percentage of
remaining live HSCs was similar with or without TGFα-PE38 (106 and
100%, respectively; Fig. 3).
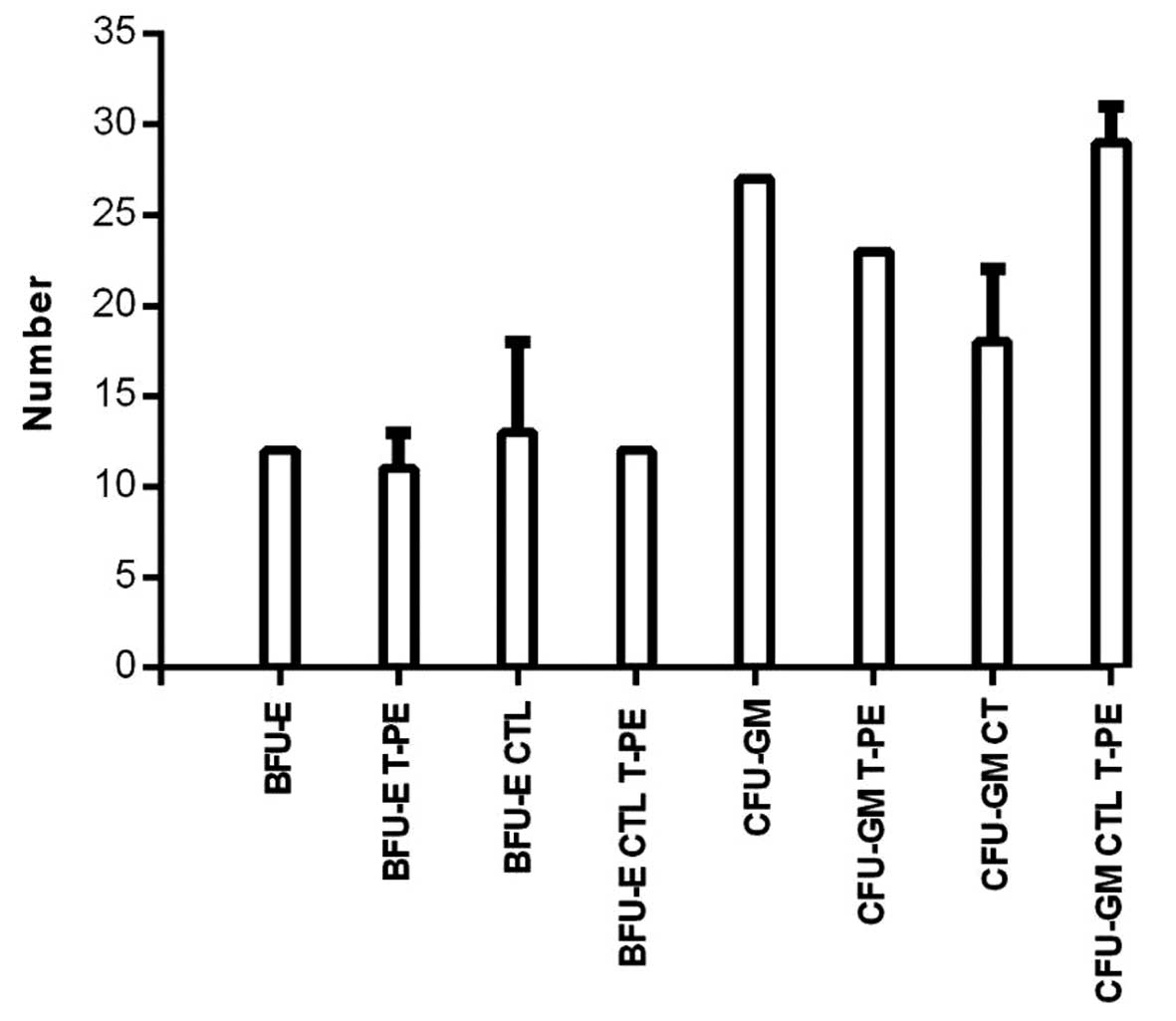
Effect of TGFα-PE38 and/or CTLs on HSC
growth and differentiation
To determine if TGFα-PE38 and/or CTLs were
inhibitory for growth and differentiation of normal cells, frozen
human peripheral blood HSCs were thawed and incubated with or
without 5 ng/ml TGFα-PE38 and/or CTLs for 14 days. The 14-day
incubation time was selected since this is the time required to see
macroscopic colonies in order to assay for BFU-E and CFU-GM
(19). The mean number of BFU-E and
CFU-GM were not significantly different with or without TGFα-PE38
(BFU-E with, 11, and without, 12; and CFU-GM with, 23, and without,
27) or for CTLs with or without TGFα-PE38 (BFU-E with, 12, and
without, 13; and CFU-GM with, 29, and without, 18) (Fig. 4).
Effect of TGFα-PE38 on CTL cell
viability
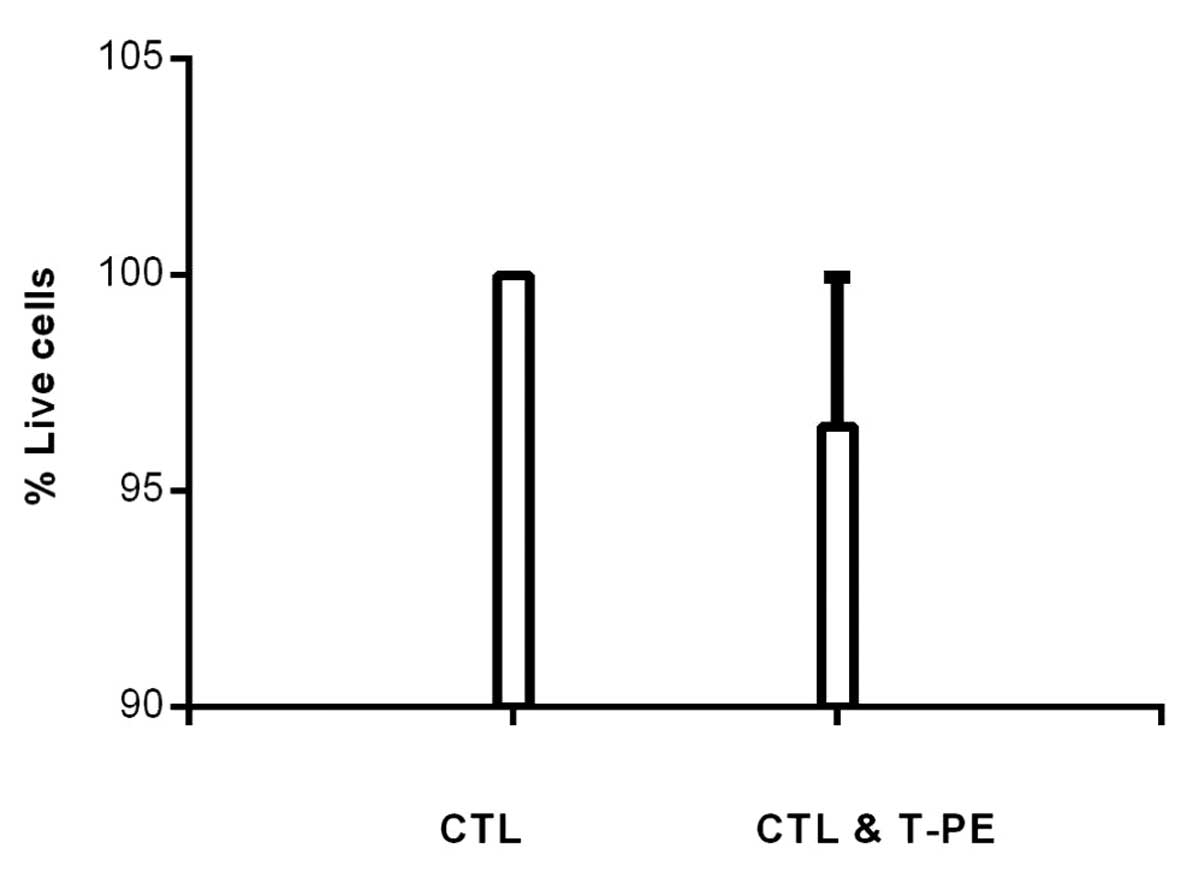
To determine if TGFα-PE38 was non-toxic for CTLs,
CTLs were incubated with or without 5 ng/ml TGFα-PE38 for three
days. The three-day incubation time was selected, as we had shown
that the majority of the MCF7 cells were killed by TGFα-PE38 at 72
h and that the lytic function of CTLs was reduced with time in
culture (6). The percentage of
remaining live CTLs was similar with or without TGFα-PE38 (97 and
100%, respectively; Fig. 5).
Effect of TGFα-PE38 on the killing
function of CTLs
To determine if TGFα-PE38 inhibited the killing
function of CTLs, CTLs were incubated with or without TGFα-PE38 for
one day. The one-day preincubation time was selected, as the lytic
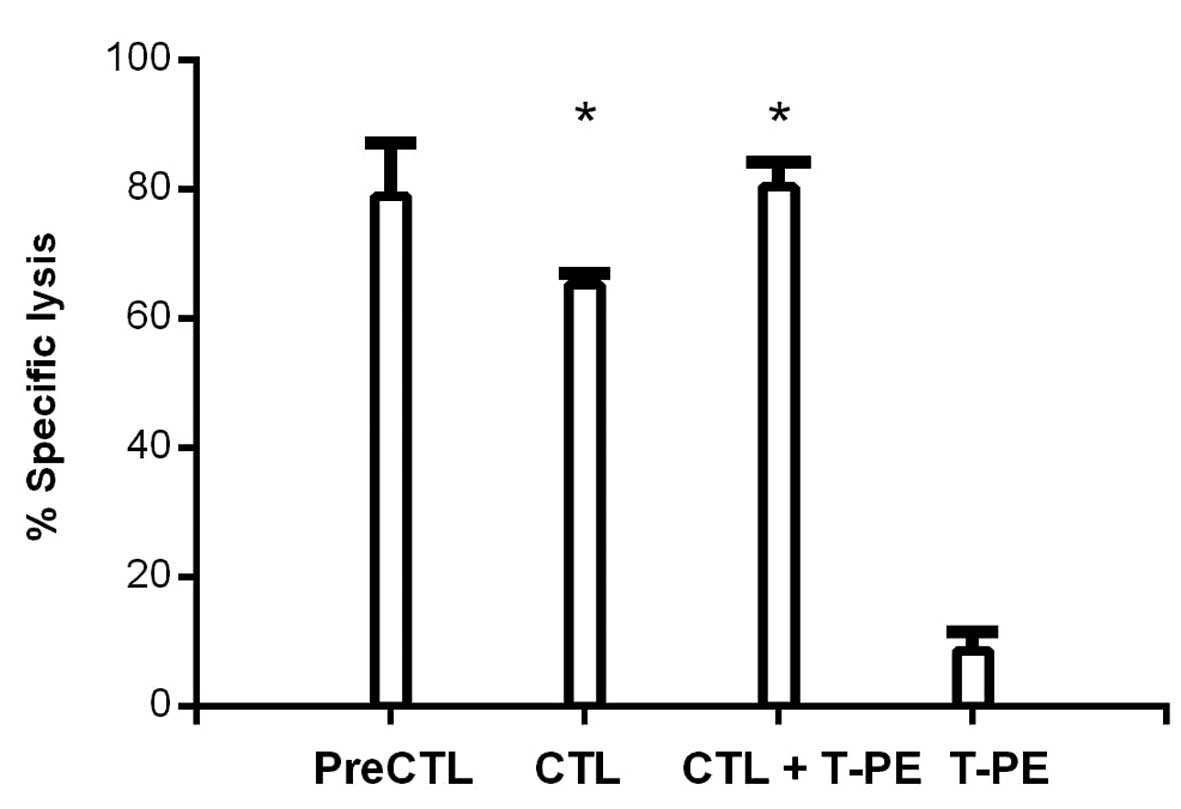
function of CTLs is reduced with time in culture (6). The %SL of the MCF7 cells by CTLs was
not inferior following preincubation with TGFα-PE38 (79%) versus
without TGFα-PE38 (65%) (Fig. 6).
To determine if TGFα-PE38 enhanced the specific lysis of the MCF7
cells by CTLs, TGFα-PE38 was co-incubated with CTLs in a one-day
specific lysis assay of the MCF7 cells. The one-day incubation time
was selected, as this is the standard time for the MCF7 specific
lysis assay (6). The concentration
of TGFα-PE38 was lowered to obtain a specific lysis of the MCF7
cells of 9%, so that an additive effect of CTLs could be observed.
Co-incubation with CTLs and TGFα-PE38 produced additive effects in
the specific lysis of the MCF7 cells (80%) (Fig. 6). This result was significantly
different (P=0.01) to CTLs alone (65%). The specific lysis of the
MCF7 cells following co-incubation with CTLs and TGFα-PE38 (80%)
was not significantly different (P=0.22) to CTLs preincubated with
TGFα-PE38 (79%) (Fig. 6).
Discussion
We aimed to affirm or refute whether the combination
of two modalities of immunotherapy, targeting two different tumor
antigens, may be feasible and non-toxic, yet enhance killing of a
human breast cancer cell line. The first target selected was EGFR,
since it is present on multiple types of tumors (1). Since TGFα is similar in structure to
EGF and binds to EGFR at a similar rate as EGF (3), it was selected as the ligand for EGFR.
Additionally, since fusion of a toxin to a ligand for the receptor
produces a specifically targeted cytotoxic agent (1), a ligand, TGFα, fused to a toxin, PE,
was used (1). TGFα-PE38 has been
shown to be cytotoxic to tumor cells in vitro (4). In addition, TGFα-PE38 reduces the size
of human brain tumor cell lines in athymic, nude mice (5). As predicted from these previous
studies, TGFα-PE38 was shown to lyse a human breast cancer cell
line, MCF7, in a concentration- and time-dependent manner. In order
for a product to be used in humans it must be non-toxic for normal
cells. Therefore, TGFα-PE38 was evaluated for toxicity against
normal cells. It was non-toxic for normal cells, specifically for
frozen human peripheral blood HSCs. Furthermore, TGFα-PE38 did not
inhibit the function of peripheral blood HSCs. The second targeted
therapy was M1SMC, which produce CTLs. We previously demonstrated
that these kill human breast cancer cells in vitro (6), and prevent human breast cancer cell
tumor development in vivo in NOD-SCID mice (7). TGFα-PE38 was not only non-toxic to
CTLs, but it also did not inhibit the specific lysis of a human
breast cancer cell line by CTLs, either as a preincubation or in
co-incubation. Instead, TGFα-PE38 enhanced the specific lysis of a
human breast cancer cell line by CTLs. These results support the
enhancement of tumor cell killing using two targets for
immunotherapy, as we have previously shown with two different
targets for developing CTLs (20).
In summary, the targeting of two tumor antigens by
two different immunotherapeutic modalities was shown to be
feasible, non-toxic and superior to either of the individual
modalities in the specific lysis of a human breast cancer cell
line. The combination of these two modalities of therapy, targeting
two different tumor antigens, may be of use in humans in preventing
recurrence of breast cancer following autologous hematopoietic stem
cell transplantation (21,22), by purging the contaminating breast
cancer cells, which are associated with recurrence (23), from the hematopoietic stem cells. In
addition, these two modalities of immunotherapy may be of benefit
in vivo for humans with breast cancer with or without other
therapies, including following autologous hematopoietic stem cell
transplantation.
Acknowledgements
The authors are grateful to the Coffee Memorial
Blood Center (Amarillo, TX, USA) and the Harrington Cancer Center
(Amarillo, TX, USA) for the PBMCs, and to those mentioned in the
text for materials and/or services. Gigi O’Connell participated in
the initial phase of the studies. This study was supported in part
by VA medical research funds (no. 0006), the Harrington Research
Foundation (Amarillo, TX, USA), the Women’s Health Research
Institute, Texas Tech University Health Sciences Center (Amarillo,
TX, USA) and the Intramural Research Program of the NIH, National
Cancer Institute, Center for Cancer Research.
References
|
1
|
Siegall CB, FitzGerald DJ and Pastan I:
Selective killing of tumor cells using EGF or TGF
alpha-Pseudomonas exotoxin chimeric molecules. Semin Cancer
Biol. 1:345–350. 1990.PubMed/NCBI
|
|
2
|
Siegall CB, Xu YH, Chaudhary VK, Adhya S,
Fitzgerald D and Pastan I: Cytotoxic activities of a fusion protein
comprised of TGF alpha and Pseudomonas exotoxin. FASEB J.
3:2647–2652. 1989.PubMed/NCBI
|
|
3
|
Prestrelski SJ, Arakawa T, Wu CS, O’Neal
KD, Westcott KR and Narhi LO: Solution structure and dynamics of
epidermal growth factor and transforming growth factor alpha. J
Biol Chem. 267:319–322. 1992.PubMed/NCBI
|
|
4
|
Chiron MF, Fryling CM and FitzGerald D:
Furin-mediated cleavage of Pseudomonas exotoxin-derived
chimeric toxins. J Biol Chem. 272:31707–31711. 1997.PubMed/NCBI
|
|
5
|
Phillips PC, Levow C, Catterall M, Colvin
OM, Pastan I and Brem H: Transforming growth
factor-α-Pseudomonas exotoxin fusion protein (TGF-α-PE38)
treatment of subcutaneous and intracranial human glioma and
medulloblastoma xenografts in athymic mice. Cancer Res.
54:1008–1015. 1994.
|
|
6
|
Wright SE, Khaznadar R, Wang Z, et al:
Generation of MUC1-stimulated mononuclear cells using optimized
conditions. Scand J Immunol. 67:24–29. 2008.PubMed/NCBI
|
|
7
|
Wright SE, Rewers-Felkins KA, Quinlin IS,
et al: Adoptive immunotherapy of mucin1 expressing adenocarcinomas
with mucin1 stimulated human peripheral blood mononuclear cells.
Int J Mol Med. 9:401–404. 2002.PubMed/NCBI
|
|
8
|
Quinlin IS, Burnside JS, Dombrowski KE,
Phillips CA, Dolby N and Wright SE: Context of MUC1 epitope:
immunogenicity. Oncol Rep. 17:453–456. 2007.PubMed/NCBI
|
|
9
|
Wright SE, Kilinski L, Talib S, et al:
Cytotoxic T lymphocytes from humans with adenocarcinomas stimulated
by native MUC1 mucin and a mucin peptide mutated at a glycosylation
site. J Immunother. 23:2–10. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wright SE, Rewers-Felkins KA, Quinlin IS,
et al: MHC-unrestricted lysis of MUC1-expressing cells by human
peripheral blood mononuclear cells. Immunol Invest. 37:215–225.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alajez NM, Schmielau J, Alter MD, Cascio M
and Finn OJ: Therapeutic potential of a tumor-specific,
MHC-unrestricted T-cell receptor expressed on effector cells of the
innate and the adaptive immune system through bone marrow
transduction and immune reconstitution. Blood. 105:4583–4589. 2005.
View Article : Google Scholar
|
|
12
|
Kanof ME and Smith PD: Preparation of
human mononuclear cell populations and subpopulations. Current
Protocols in Immunology. Coligan JE: John Wiley & Sons, Inc;
New York, NY: pp. 72009
|
|
13
|
Barnd DL, Lan MS, Metzgar RS and Finn OJ:
Specific, major histocompatibility complex-unrestricted recognition
of tumor-associated mucins by human cytotoxic T cells. Proc Natl
Acad Sci USA. 86:7159–7163. 1989. View Article : Google Scholar
|
|
14
|
Roehm NW, Rodgers GH, Hatfield SM and
Glasebrook AL: An improved colorimetric assay for cell
proliferation and viability utilizing the tetrazolium salt XTT. J
Immunol Methods. 142:257–265. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jost LM, Kirkwood JM and Whiteside TL:
Improved short- and long-term XTT-based colorimetric cellular
cytotoxicity assay for melanoma and other tumor cells. J Immunol
Methods. 147:153–165. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Altman SA, Randers L and Rao G: Comparison
of Trypan blue dye exclusion and fluorometric assays for mammalian
cell viability determinations. Biotechnol Prog. 9:671–674. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bode AM, Ma W-Y, Surh Y-J and Dong Z:
Inhibition of epidermal growth factor-induced cell transformation
and activator protein 1 activation by [6]-gingerol. Cancer Res.
61:850–853. 2001.
|
|
18
|
Vennstrom L, Bysell C, Bjorkelund H,
Lundqvist H and Andersson K: Real-time viability assay based on
51Cr retention in adherent cells. Biotechniques. 44:237–240. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu MH, Liebowitz DN, Smith SL, Williams SF
and Dolan ME: Efficient expression of foreign genes in human CD34+
hematopoietic precursor cells using electroporation. Gene Therapy.
8:3842001.
|
|
20
|
Petty AP, Wright SE, Rewers-Felkins KA,
Yenderrozos MA, Vorderstrasse BA and Lindsey JS: Targeting
migration inducting gene-7 inhibits carcinoma cell invasion, early
primary tumor growth, and stimulates monocyte oncolytic activity.
Mol Cancer Ther. 8:2412–2423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ross AA, Cooper BW, Lazarus HM, et al:
Detection and viability of tumor cells in peripheral blood stem
cell collections from breast cancer patients using
immunocytochemical and clonogenic assay techniques. Blood.
82:2605–2610. 1993.PubMed/NCBI
|
|
22
|
Rill DR, Santana VM, Roberts WM, et al:
Direct demonstration that autologous bone marrow transplantation
for solid tumors can return a multiplicity of tumorigenic cells.
Blood. 84:380–383. 1994.PubMed/NCBI
|
|
23
|
Braun S, Vogl FD, Naume B, et al: A pooled
analysis of bone marrow micrometastasis in breast cancer. N Engl J
Med. 353:793–802. 2005. View Article : Google Scholar : PubMed/NCBI
|