Introduction
Lymphomatoid granulomatosis (LYG) is a rare
Epstein-Barr virus (EBV)-associated disorder and is considered part
of the spectrum of lymphoproliferative disorders. While commonly
presenting with multiple lung nodules, in occasional cases,
atypical patterns can be observed radiographically. Specific
systemic manifestations can occur, including fever, weight loss,
skin lesions and the involvement of other organs (1). The clinical presentation of LYG can
mimic infectious diseases (including tuberculosis), vasculitis or
metastatic malignancies. According to the 2008 WHO classification,
LYG is characterized by the proliferation of B-lymphoma cells
(2). Patients may present with
non-specific neurological symptoms, including seizures and
incontinence (3). In order to make
a definitive diagnosis, biopsies, pathological examinations and
immunohistochemical analyses must be performed. In the current
study, an overview of the literature in reference to the etiology,
clinical features, diagnosis and treatment options for LYG is
presented. Patient provided written informed consent.
Case report
A 19-year-old male was presented to the pulmonary
clinic of the People’s hospital of Shenzhen University (Shenzhen,
China) with chills, a fever, mild intermittent dry coughing, night
sweating and fatigue that had lasted 7 days. One month prior to
this presentation, the illness began with a low fever, mild
coughing and fatigue. The patient was treated symptomatically for a
presumed upper-respiratory viral infection. The patient had fevers
as high as 40.2°C and had no significant exposure history. The
patient had no risk factors for human immunodeficiency virus (HIV)
infection, and was an ex-smoker.
Upon presentation, the patient did not experience
respiratory distress and the vital signs were as follows:
Temperature, 40.2°C; heart rate, 110 beats/min; respiratory rate,
24 breaths/min; and blood pressure, 120/70 mmHg. Oxygen saturation
(measured via digital pulse oximetry) was 95% while breathing
ambient air. The skin was diaphoretic and there were no palpable
lymph nodes in all areas. Chest, cardiac, abdominal and skin
examinations did not reveal abnormalities. On lung auscultation,
breath sounds were diminished over the lower left lung, and there
were no crackles or rales on either side. A neurological
examination revealed no focal defect.
The white blood cell count of the patient was
4.7×103/dl and the hemoglobin level was 13.0 g/dl. Serum
electrolyte levels and liver and renal function were normal.
Spirometry values and the erythrocyte sedimentation rate (ESR; 7
mm/h) were normal. Routine blood and sputum cultures for detection
of acid-fast bacteria were negative. Serological tests revealed
that there was no infection with hepatitis B virus, hepatitis C
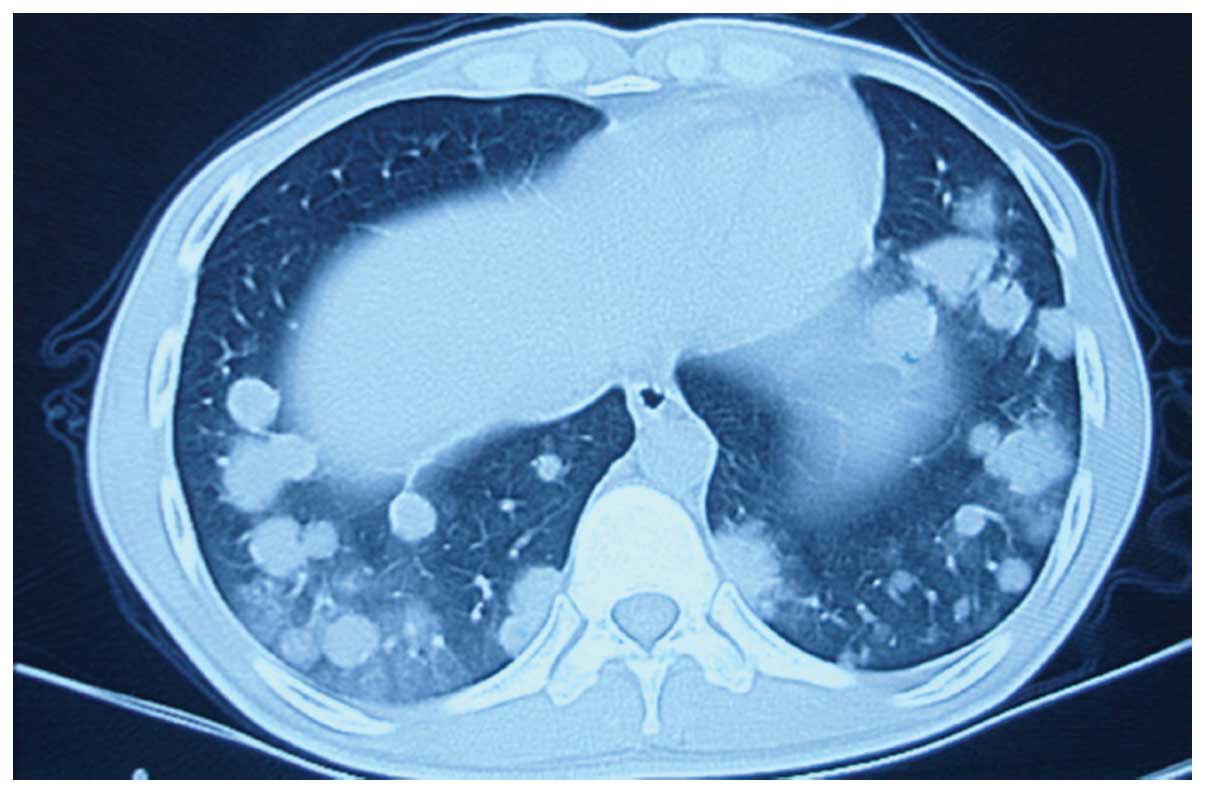
virus, HIV and tubercle bacillus. Computed tomography (CT) imaging
demonstrated bilateral pulmonary multiple round nodules,
predominantly in the lower lung fields, along the bronchoalveolar
structures (Fig. 1). There was no
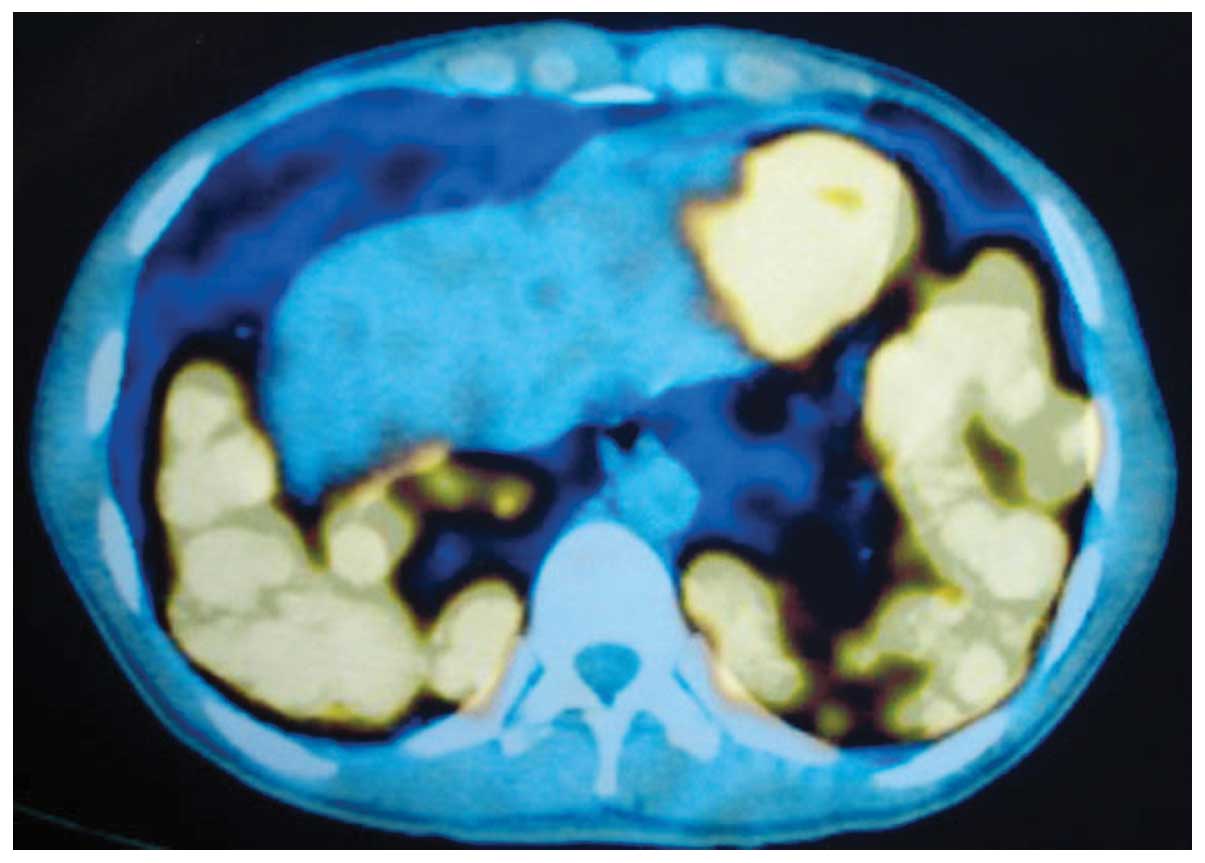
mediastinal or hilar lymph node enlargement. F-18
fluorodeoxyglucose (FDG) positron emission tomography (PET)
revealed multiple hypermetabolic pulmonary nodules in the lungs,
predominantly in the lower lobes [maximum standard uptake value
(SUVmax), 3.3–7.8; Fig. 2).
Splenomegaly was observed and the SUVmax was 12.4 in the PET/CT
image. The CT-guided transthoracic biopsy revealed inflammatory
cells and large coagulated necrotic tissue. The patient consented
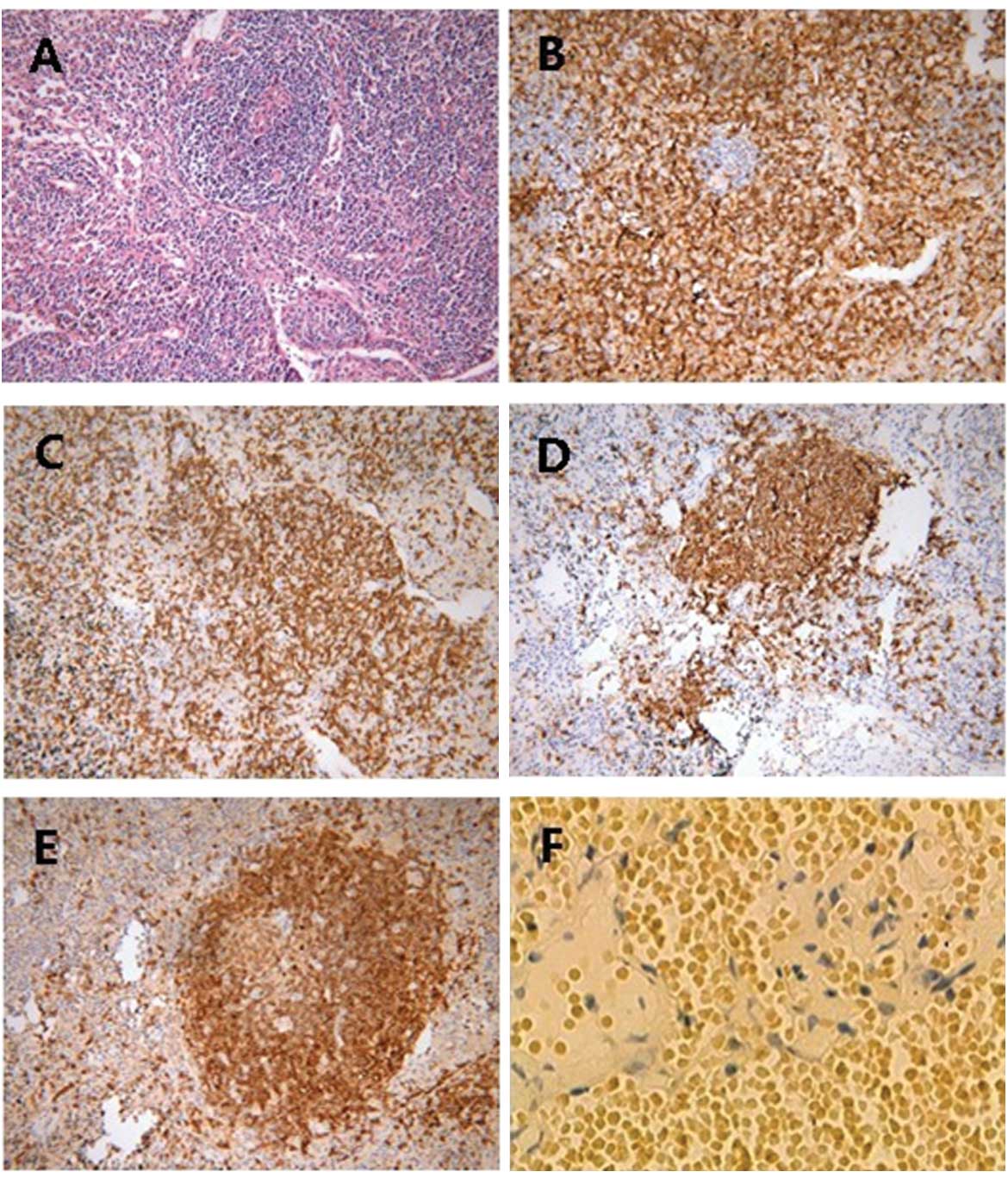
to the removal of a thoracoscopic biopsy, which revealed an
angiocentric and angiodestructive infiltrate of lymphoid cells in
the vascular wall, with surrounding infarct-like tissue necrosis
(Fig. 3). Immunohistochemical
staining revealed that the lymphoid infiltrate consisted of a
mixture of cells expressing cluster of differentiation (CD)45RO,
CD3, CD20 and CD79α. Furthermore, in situ hybridization for
the detection of EBV was strongly positive (Fig. 3). A diagnosis of LYG was finally
established.
The patient received chemotherapy with
cyclophosphamide, adriamycin, vincristine and prednisone (CHOP).
The body temperature was normal during the chemotherapy. However,
only two days subsequent to the end of chemotherapy, the patient
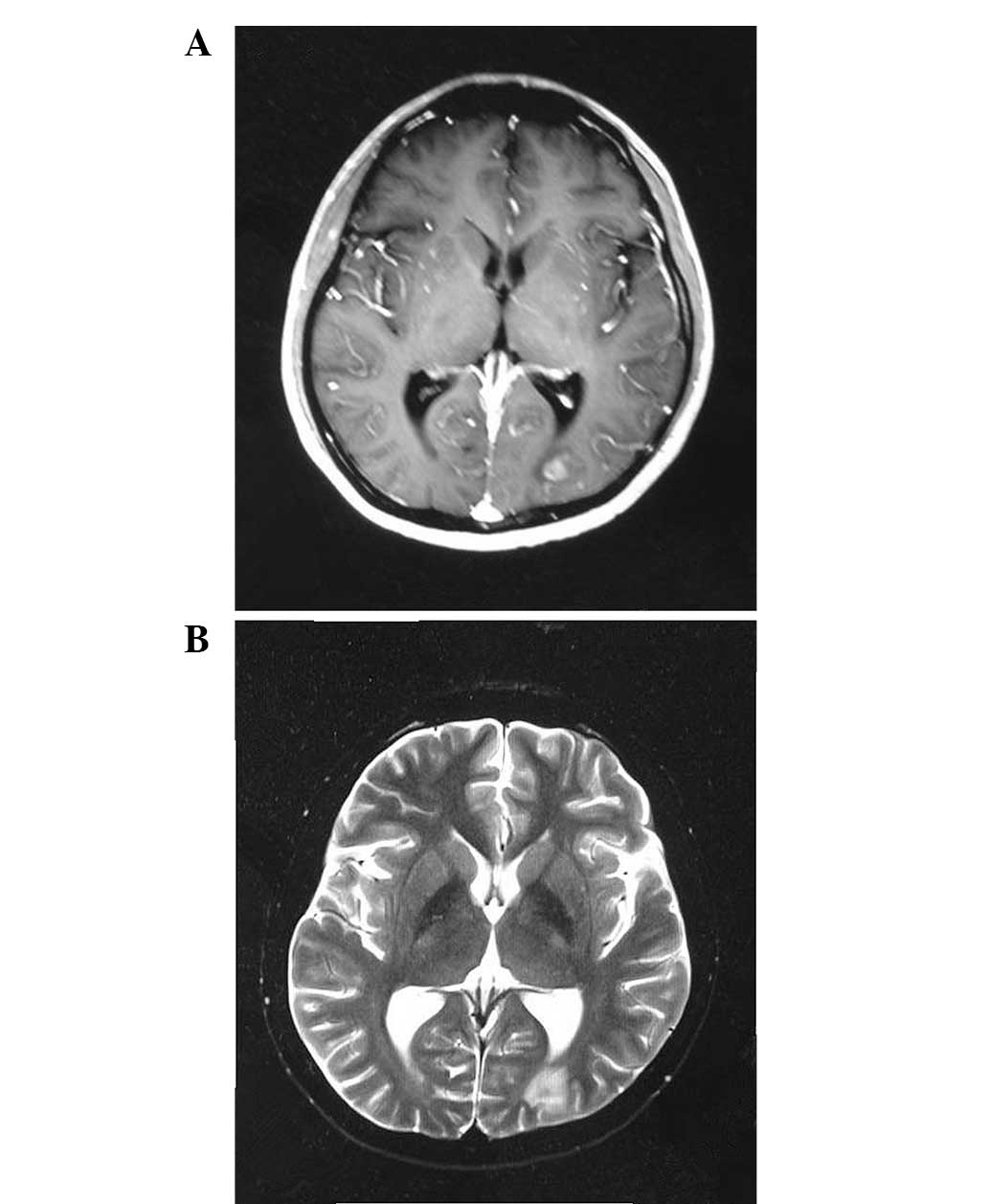
developed a generalized tonic-clonic seizure and fever. T1- and
T2-weighted magnetic resonance (MR) images revealed multiple
irregular and nodular lesions, with peripheral edema in the
bilateral parietal lobe and the left cornu occipitale of the
substantia alba (Fig. 4). Central
nervous system (CNS) involvement was considered. Therefore, the
patient received whole brain irradiation with a total dose of 30 Gy
in 10 fractions. This treatment did not halt the generalized
tonic-clonic seizure, while the coughing, shortness of breath and
hyperpyrexia continued. A chest-CT revealed a left-sided pleural
effusion, lobus inferior pulmonis atelectasis and small amounts of
pericardial effusion. The patient received 2 l/min oxygen by nasal
canula. Subsequently, the patient developed rapid pulmonary
failure, and succumbed ~3 months after presentation due to
progressive pulmonary and CNS disease.
Discussion
LYG is a rare lymphoproliferative disease. Liebow
et al designated the term ‘lymphomatoid granulomatosis’
almost 40 years ago, as an angiocentric and angiodestructive
lymphoreticular proliferative and granulomatous disease with EBV
infection, lung lesions and a poor outcome (4). The first description of the disease
was followed by several reports about the nature, histopathology
and treatment of LYG. The following is an overview of this
literature.
LYG has an increased male predilection, with
male-female ratios ranging between 2:1 and 3:1. Generally, patients
are between 40 and 60 years old (5). LYG frequently presents as a systemic
disease, usually with prominent pulmonary sites. Other sites can
also be involved, including the skin, CNS, kidney, liver, upper
respiratory tract and gastrointestinal tract. Lymph node and spleen
involvement are less common (7–8%). Constitutional symptoms,
including weight loss, fatigue, sweating and chills, are typical
(6). Local symptoms include
coughing, shortness of breath and chest pain, while almost
one-third of patients develop neurological symptoms, including
confusion, ataxia, hemiparesis or seizures, mainly due to lesions
in the CNS. Cranial nerve palsies and peripheral polyneuropathy
have also been described in 7% of cases (7–10).
Chest X-rays reveal bilateral lesions in 71–90% of
patients. The most frequent observations are multiple nodules or
masses with poorly defined margins, whereas reticular or nodular
infiltrates are less often described. Cavitations or solitary
masses are rarely presented. Pleural effusions (25%) are usually
small and present as costophrenic angle blunting. Mediastinal
lymphadenopathy is visible on CT in 60% of patients, and nodules
are characteristically observed alongside bronchovascular
structures and interlobular septa (11,12).
The application of FDG-PET for tumor imaging has proved to be
highly useful for the diagnosis of primary lung tumors. If LYG is
radiologically proven to involve the CNS, this is shown as multiple
punctate and linear enhancement on CT and MR images, representing
lesions in perivascular tissue and the walls of small blood vessels
(13). Patsalides et al
(14) conducted a comprehensive
retrospective analysis of the MRI results of 25 patients with LYG
and found a wide spectrum of CNS lesions in 52% (13/25) of the
patients. In the most frequent cases, the patients presented with
multifocal intraparenchymal brain lesions, which exhibited punctate
or linear enhancement (n=7), followed by leptomeningeal and cranial
nerve involvement (n=6).
LYG is similar to Wegener’s granulomatosis in
clinical features and imaging, including pulmonary manifestations
and the systemic nature. The differential diagnosis includes
tuberculosis, granulomatosis, lung cancer and inflammatory
pseudotumors. The prognosis of LYG is extremely variable, ranging
from extremely good in the cases of spontaneous resolution, to
rapidly fatal. The median survival time ranges between 14 months
and 4 years, and the 5-year mortality rate is 60–90%. In total, 94%
of mortalities occur in the first 36 months following diagnosis
(15–17). Certain studies indicate that
leucopenia, fever, anergy in reaction to common skin test antigens,
a young age and localization in the CNS are poor prognostic signs
(7,8,18). A
biopsy must be taken to make a definitive diagnosis. Transbronchial
biopsy is not recommended, as it is diagnostic in only 27% of
cases, while open lung biopsy specimens are uniformly positive
(19).
As LYG is a rare disease, a standard treatment
paradigm has not yet been established, and consequently, the
treatment of LYG remains a challenge. Certain patients have
self-limiting disease, a number are treated with corticosteroids,
either as single agent or combined with cyclophosphamide, and
certain patients are treated with another chemotherapy, including
CHOP or COP regimens, depending on severity at presentation. LYG
responds poorly to conventional chemotherapy, and no significant
differences have been identified between the regimens, while
radiotherapy has also been used for CNS and orbital localizations
(7,20,21).
Rituximab-CHOP, or other similar regimens, are usually recommended
for treatment of grade 3 LYG due to the high rate of rapid
progression into EBV-positive large B-cell lymphoma (22,23).
Several case studies have been published in which treatments with
interferon α-2b, with or without autologous stem cell
transplantation (24–26), and prednisone, methotrexate,
doxorubicin, cyclophosphamide, etoposide, mechlorethamine,
vincristine, and procarbazine or cyclosporin-A were presented
(27). For the patient in the
present case study, CHOP chemotherapy and radiotherapy failed, and
rapid progressive disease was observed in the CNS. The present case
demonstrated that LYG is a chemotherapy-resistant disease in
certain patients. LYG remains rarely recognized as an entity, and
proper diagnosis and treatment remain a challenge.
Acknowledgements
This project was supported by the Natural Science
Foundation of China (no. 30900597) and the Science Foundation of
Hubei Health Department (no. JX6B08).
References
|
1
|
de Boysson H and Geffray L: Lymphomatoid
granulomatosis. Rev Med Interne. 34:349–357. 2013.(In French).
|
|
2
|
Vardiman JW: The World Health Organization
(WHO) classification of tumors of the hematopoietic and lymphoid
tissues: An overview with emphasis on the myeloid neoplasms. Chem
Biol Interact. 184:16–20. 2010. View Article : Google Scholar
|
|
3
|
Dister F and Ghaye B: Lymphomatoid
granulomatosis. JBR-BTR. 95:140–141. 2012.PubMed/NCBI
|
|
4
|
Liebow AA, Carrington CR and Friedman PJ:
Lymphomatoid granulomatosis. Hum Pathol. 3:457–558. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mazzie JP, Price AP, Khullar P, et al:
Lymphomatoid granulomatosis in a pediatric patient. Clin Imaging.
28:209–213. 2004. View Article : Google Scholar
|
|
6
|
Lundell RB, Weenig RH and Gibson LE:
Lymphomatoid granulomatosis. Cancer Treat Res. 142:265–272.
2008.PubMed/NCBI
|
|
7
|
Katzenstein AL, Carrington CB and Liebow
AA: Lymphomatoid granulomatosis: a clinicopathologic study of 152
cases. Cancer. 43:360–373. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fauci AS, Haynes BF, Costa J, Katz P and
Wolff SM: Lymphomatoid granulomatosis. Prospective clinical and
therapeutic experience over 10 years. N Engl J Med. 306:68–74.
1982.PubMed/NCBI
|
|
9
|
Koss MN, Hochholzer L, Langloss JM, Wehunt
WD, Lazarus AA and Nichols PW: Lymphomatoid granulomatosis: a
clinicopathologic study of 42 patients. Pathology. 18:283–288.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Katzenstein AL and Peiper SC: Detection of
Epstein-Barr virus genomes in lymphomatoid granulomatosis: analysis
of 29 cases by the polymerase chain reaction technique. Mod Pathol.
3:435–441. 1990.PubMed/NCBI
|
|
11
|
Wechsler RJ, Steiner RM, Israel HL and
Patchefsky AS: Chest radiograph in lymphomatoid granulomatosis:
comparison with Wegener granulomatosis. AJR Am J Roentgenol.
142:79–83. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JS, Tuder R and Lynch DA: Lymphomatoid
granulomatosis: radiologic features and pathologic correlations.
AJR Am J Roentgenol. 175:1335–1339. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tateishi U, Terae S, Ogata A, et al: MR
imaging of the brain in lymphomatoid granulomatosis. AJNR Am J
Neuroradiol. 22:1283–1290. 2001.PubMed/NCBI
|
|
14
|
Patsalides AD, Atac G, Hedge U, et al:
Lymphomatoid granulomatosis: abnormalities of the brain at MR
imaging. Radiology. 237:265–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jaffe ES and Wilson WH: Lymphomatoid
granulomatosis: pathogenesis, pathology and clinical implications.
Cancer Surv. 30:233–248. 1997.PubMed/NCBI
|
|
16
|
Cadranel J, Wislez M and Antoine M:
Primary pulmonary lymphoma. Eur Respir J. 20:750–762. 2002.
View Article : Google Scholar
|
|
17
|
Katherine Martin L, Porcu P, Baiocchi RA,
Erter JW and Chaudhury AR: Primary central nervous system
lymphomatoid granulomatosis in a patient receiving azathioprine
therapy. Clin Adv Hematol Oncol. 7:65–68. 2009.PubMed/NCBI
|
|
18
|
Wilson WH, Kingma DW, Raffeld M, Wittes RE
and Jaffe ES: Association of lymphomatoid granulomatosis with
Epstein-Barr viral infection of B lymphocytes and response to
interferon-alpha 2b. Blood. 87:4531–4537. 1996.PubMed/NCBI
|
|
19
|
Pisani RJ and DeRemee RA: Clinical
implications of the histopathologic diagnosis of pulmonary
lymphomatoid granulomatosis. Mayo Clin Proc. 65:151–163. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sordillo PP, Epremian B, Koziner B, Lacher
M and Lieberman P: Lymphomatoid granulomatosis: an analysis of
clinical and immunologic characteristics. Cancer. 49:2070–2076.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Petrella TM, Walker IR, Jones GW and Leber
B: Radiotherapy to control CNS lymphomatoid granulomatosis: a case
report and review of the literature. Am J Hematol. 62:239–241.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rao R, Vugman G, Leslie WT, Loew J and
Venugopal P: Lymphomatoid granulomatosis treated with rituximab and
chemotherapy. Clin Adv Hematol Oncol. 1:658–660. 2003.PubMed/NCBI
|
|
23
|
Ishiura H, Morikawa M, Hamada M, et al:
Lymphomatoid granulomatosis involving central nervous system
successfully treated with rituximab alone. Arch Neurol. 65:662–665.
2008. View Article : Google Scholar
|
|
24
|
Richter C, Schnabel A, Müller KM, Reuter
M, Schuster P and Gross WL: Lymphomatoid granulomatosis - remission
induction with interferon-alpha 2b. Dtsch Med Wochenschr.
122:1106–1110. 1997.(In German).
|
|
25
|
Lemieux J, Bernier V, Martel N and Delage
R: Autologous hematopoietic stem cell transplantation for
refractory lymphomatoid granulomatosis. Hematology. 7:355–358.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johnston A, Coyle L and Nevell D:
Prolonged remission of refractory lymphomatoid granulomatosis after
autologous hemopoietic stem cell transplantation with
post-transplantation maintenance interferon. Leuk Lymphoma.
47:323–328. 2006. View Article : Google Scholar
|
|
27
|
Raez LE, Temple JD and Saldana M:
Successful treatment of lymphomatoid granulomatosis using
cyclosporin-A after failure of intensive chemotherapy. Am J
Hematol. 53:192–195. 1996. View Article : Google Scholar : PubMed/NCBI
|