Introduction
Peripheral T-cell lymphomas (PTCLs) are a diverse
group of mature neoplasms that account for 12% of non-Hodgkin’s
lymphomas (NHLs) (1). They are a
rare and heterogeneous group of disorders that are predominantly
associated with a poor prognosis (2). The standard PTCL treatment is
administration of cyclophosphamide, doxorubicin, vincristine and
prednisone (CHOP) or a regimen comparable with CHOP that includes
anthracyclines. Over the last few decades, an association between
NHLs and autoimmune diseases has been gradually established
(3). Certain studies have
demonstrated that autoimmune diseases are associated with a higher
risk of developing NHLs, while other findings show that the
correlation between autoimmunity and lymphoproliferative tumors is
bi-directional. It is hypothesized that the impaired regulation of
auto-reactive or transformed lymphocytes (or a combination of the
two), via programmed cell death, leads to the development of these
two chronic conditions, which highlights the significance of the
immune system regulation via apoptosis (3).
Pemphigus is a life-threatening autoimmune
blistering disease that is classified into four major types;
pemphigus vulgaris, pemphigus foliaceus, paraneoplastic pemphigus
and immunoglobulin (Ig) A pemphigus (4). IgA pemphigus is characterized by
tissue-bound and circulating IgA antibodies, which target cell
surface components in the epidermis. This disease is
histopathologically characterized by extensive epidermal
infiltration by neutrophils and slight epidermal acantholysis. IgA
pemphigus was proposed as a disease entity relatively recently and,
to the best of our knowledge, only 60 cases have been reported to
date. The male-to-female ratio of IgA pemphigus is 1:1.33 and the
average age of the affected individuals is 53 years. IgA pemphigus
is considered to be less life-threatening than other types of
pemphigus; thus far, there have been no reports of IgA pemphigus
patients who have succumbed as a direct result of the disease
(5). The clinical manifestations of
IgA pemphigus are flaccid blisters or pustules on normal or
erythematous skin. The majority of pustules fuse and form a large
ring with scales and scabs in the center, and blisters in the
margin. Nikolsky’s sign is usually negative, however, specific
cases that exhibited positive results have been reported.
Additionally, mucous membranes are not usually affected in IgA
pemphigus.
IgA pemphigus is further subdivided into the
subcorneal pustular dermatosis (SPD)-type, whose target antigen is
desmocollin 1, and intraepidermal neutrophilic IgA dermatosis
(IEN)-type, whose target antigen is unknown although is potentially
a non-desmosomal cell surface protein (9–11). The
pustules localize in the subcorneal region in SPD-type IgA
pemphigus, whereas they are present in the entire or mid-epidermis
in IEN-type IgA pemphigus. Diagnostic direct immunofluorescence
reveals IgA deposition in the epithelial prickle cell membrane and
IgG or complement 3 (C3) may be deposited, however, to a lesser
extent than IgA. Indirect immunofluorescence typically shows
circulating IgA antibodies (7–9).
Patient provided written informed consent.
Case report
A 43-year-old male was admitted to the Union
Hospital (Wuhan, China) in March 2012 with a two-month history of
fatigue and a poor appetite. The patient also reported the presence
of a palpable mass under the jaw bone one month prior to admission
and did not report any symptoms of fever, night sweats or weight
loss. Physical examination identified multiple swollen lymph nodes
in the bilateral cervix, the supraclavicular-axillary region and
the inguinal fold. The largest node measured 4.5×3 cm2
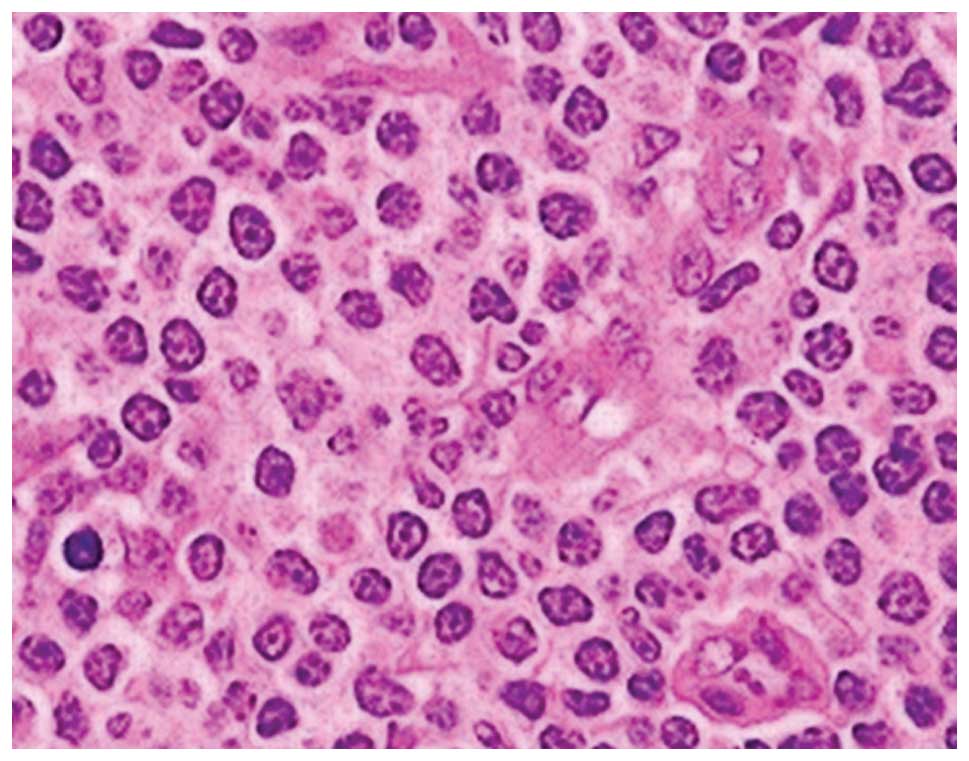
and was located in the right inguinal fold. Pathology of the right
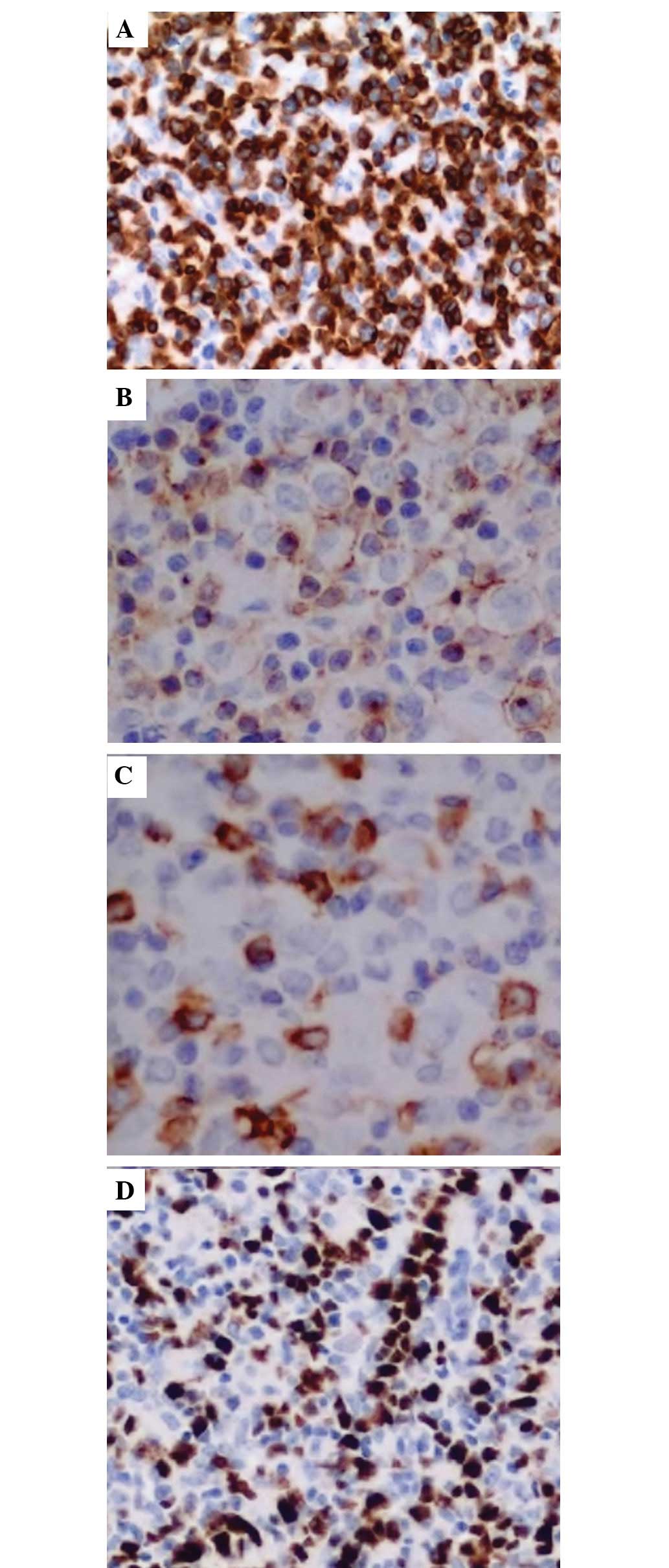
inguinal lymph node revealed PTCL. Immunohistochemical staining was
positive for cluster of differentiation (CD)2, CD3, CD5, CD7, and
CD47, and negative for CD20. Ki-67 was ~40% positive (Figs. 1 and 2A–D). The computed tomography scan
performed with a contrast agent displayed a clean mediastinum,
abdomen and pelvis, all without swollen lymph nodes. The bone
marrow exam was normal. The patient was diagnosed with PTCL and
stage IIIA disease, and received four cycles of chemotherapeutic
CHOP between March 25 and May 30, 2012; the patient response was
partial remission (PR). In June 2012, the B symptoms (lymphoma
specific symptoms: Fever, night sweats and weight loss) reported by
the patient were fever and night sweats. The lymph node in the
inguinal fold had increased in size, with a tumor evaluation
indicating progressive disease according to the Response Evaluation
Criteria in Solid Tumors (RECIST 1.1) (12). The patient received two cycles of
gemcitabine, cisplatin and dexamethasone between June 27 and July
22, 2012. The response was PR according to RECIST 1.1 (12), and the patient decided to terminate
treatment. In September, re-examination showed enlarged lymph nodes
in the retroperitoneal region, however, the other regions were
normal. The patient received a 36-Gy total dose of radiation to the
retroperitoneal region, and the response was an unconfirmed
complete remission. The patient was prescribed oral thalidomide
(125 mg daily) as maintenance therapy for one month.
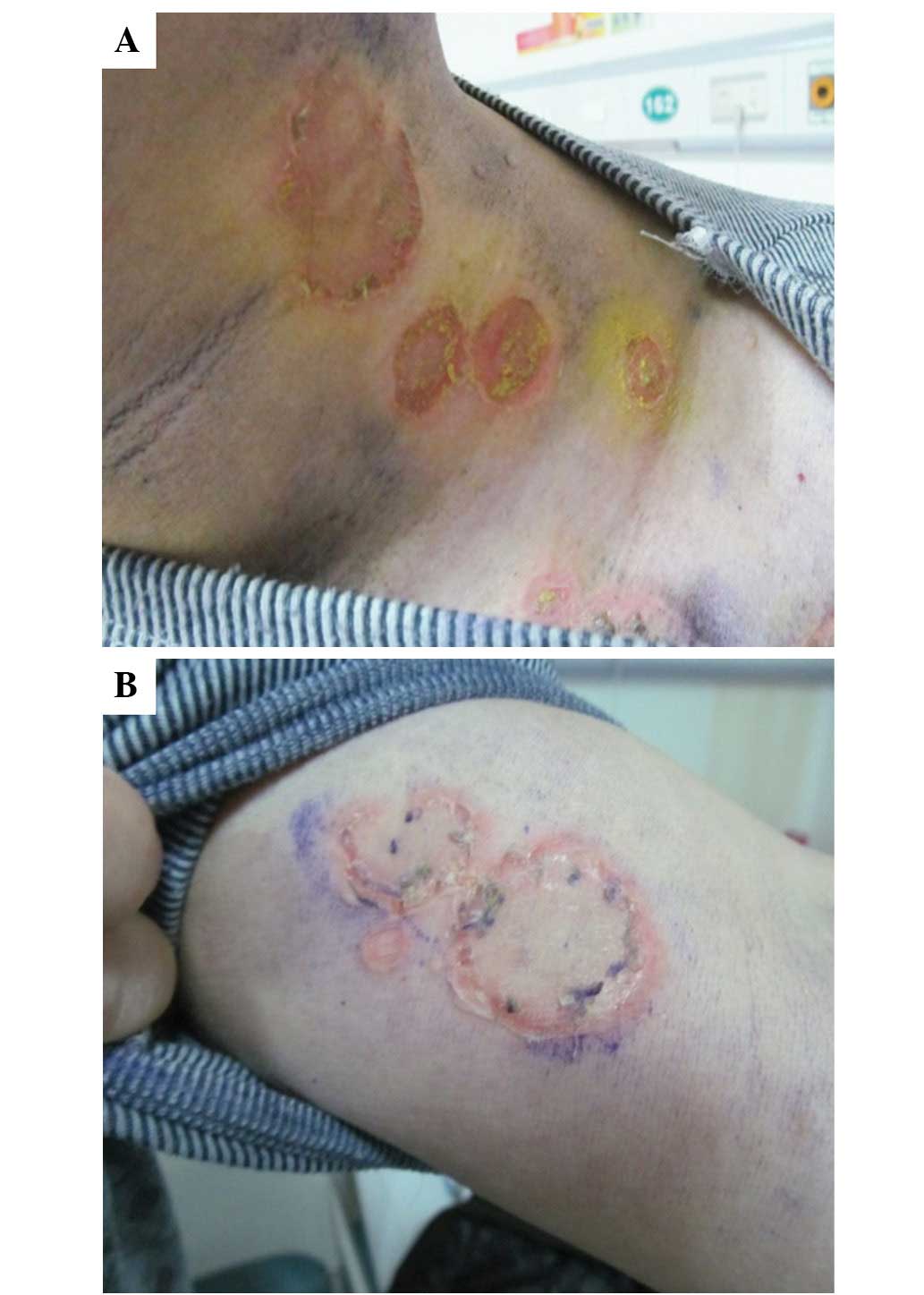
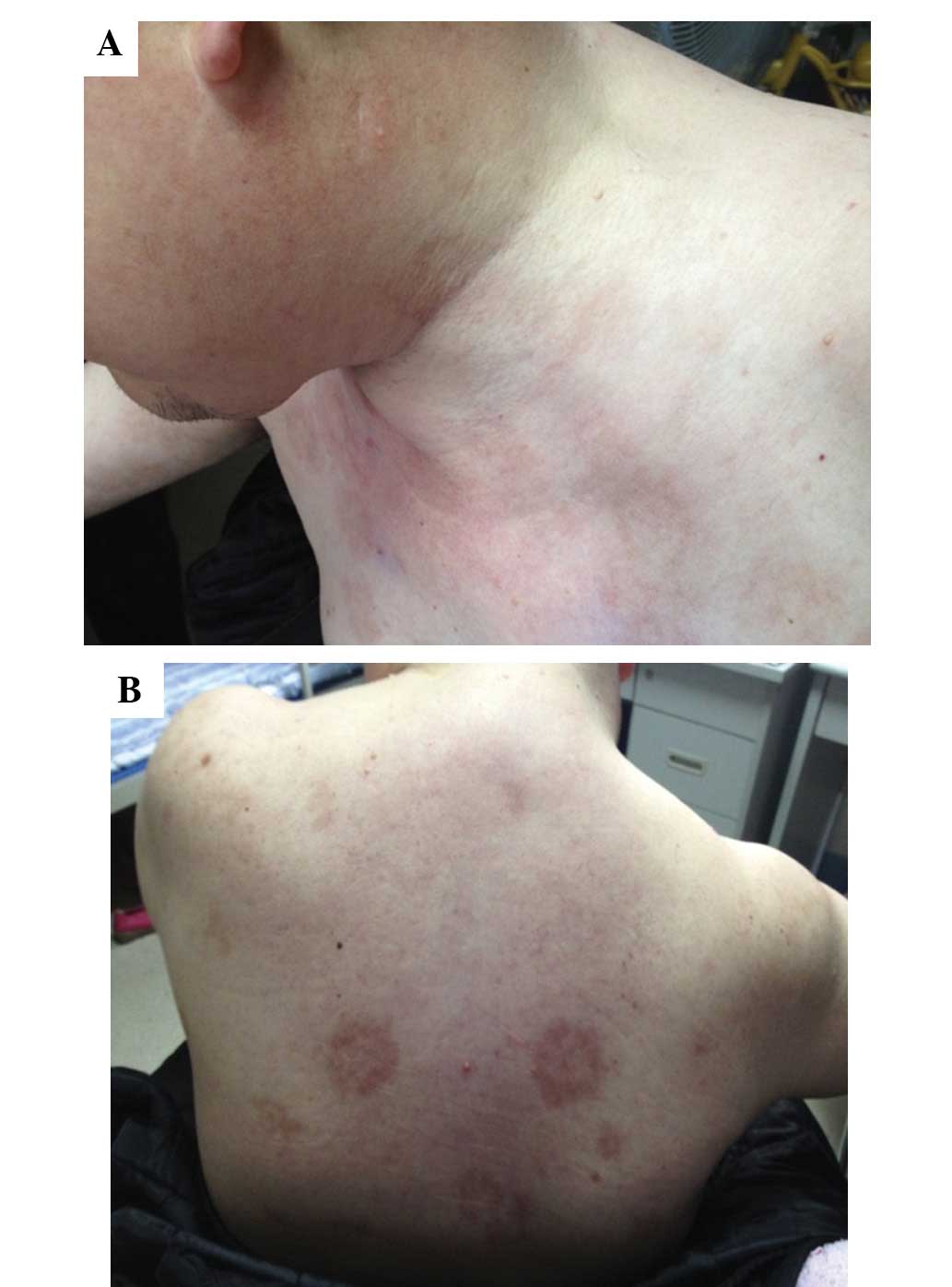
Subsequent to receiving thalidomide for ~20 days, a
number of blisters were found on the trunk and limbs of the
patient. The patient was referred to the dermatology department
where fingernail- to nut-sized eroded patches were observed on the
scalp, the four limbs, the lower abdomen and the inguinal fold. The
blisters did not exhibit obvious seepage. Nikolsky’s sign was
positive (Fig. 3A and B) and the
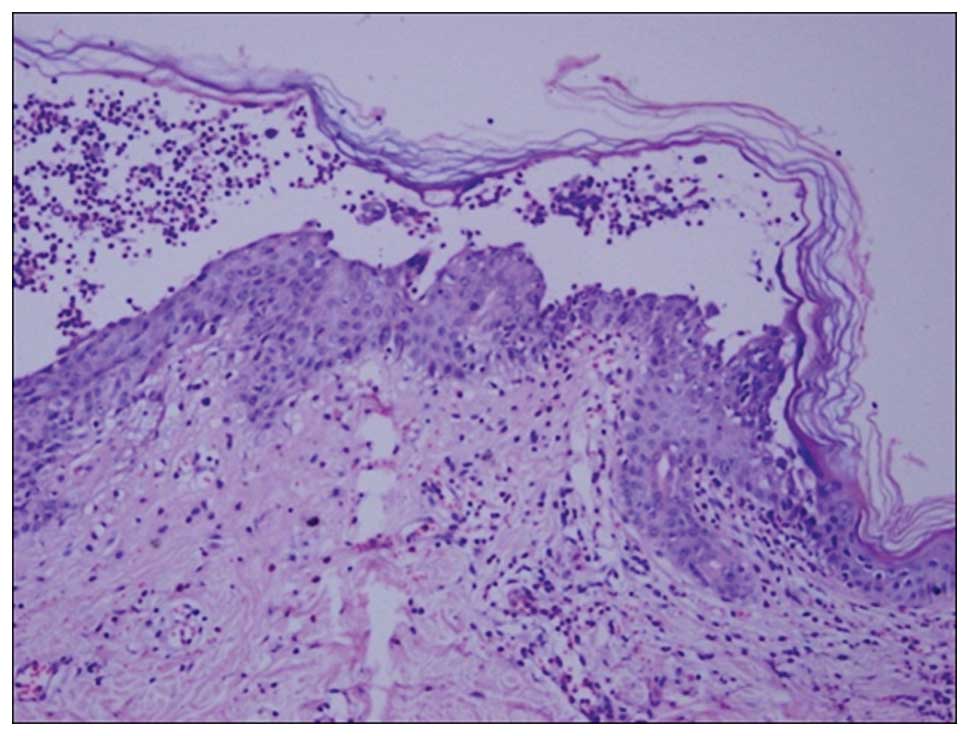
oral cavity mucous membranes were spared. Histopathology of the
skin lesion from the abdomen showed subcorneal pustular, slight
epidermal acantholysis and extensive neutrophilic infiltration in
the epidermis. Lymphocyte, histiocyte and eosinophil infiltration
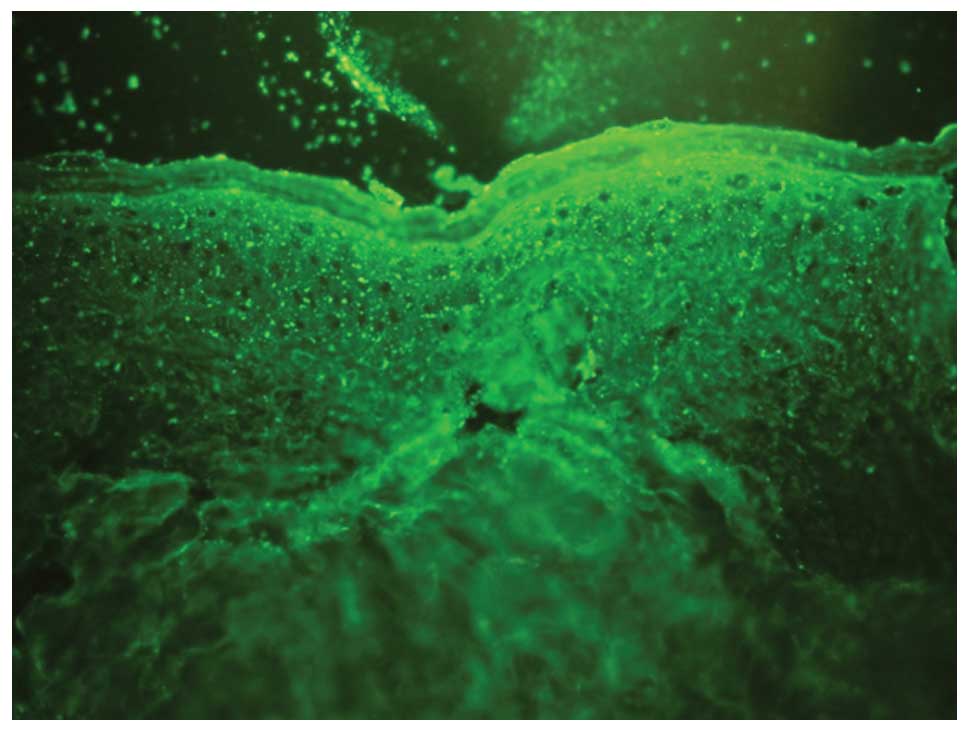
were observed in the superficial layer of the dermis (Fig. 4). Direct immunofluorescence of the
biopsy was positive for IgA and negative for IgM, IgG, C3, C4 and
C1q (Fig. 5). The clinical
manifestations, histopathology and direct immunofluorescence met
the diagnostic criteria of IgA pemphigus. Therefore, the patient
was administered with an 80-mg methylprednisolone intravenous drip
for two weeks and the blisters faded (Fig. 6). The treatment was altered to
prednisone for maintenance therapy with a starting dose of 80 mg
and was decreased by 10 mg every two weeks. The patient is
currently taking 40 mg prednisone and the lymphoma is stable. At
present, there is no evidence of recurrence of the lymphoma or
pemphigus in this patient.
Discussion
IgA pemphigus is a rare disease characterized by a
vesiculopustular eruption established by epidermal intercellular
IgA deposition. Various cases in the literature were associated
with other diseases, including IgA paraproteinemia, IgG
cryoglobulinemia, lymphoma, myeloma, rheumatoid arthritis,
ulcerative colitis, Crohn’s disease, lung cancer and
hyperthyroidism (13,14).
The chronic inflammation characteristic of IgA
pemphigus may be due to cancer progression or drug-associated
immunosuppression. Pemphigus skin diseases most often complicate
blood-lymphatic system disorders. For example, there is an
association between paraneoplastic pemphigus and NHL. One case of
chronic lymphocytic leukemia complicated by IgA pemphigus and
paraneoplastic pemphigus has been reported (15). In addition, Asahina et al
(16) reported a case of diffuse
large B cell lymphoma that was complicated by IgA pemphigus without
rash recurrence for one year after treatment.
The male patient with PTCL in the present study met
the diagnostic criteria of IgA pemphigus as a result of clinical
manifestations, histopathology and immunofluorescence assays.
Histopathology showed blistered skin, subcorneal pustules and
acantholysis, and direct immunofluorescence revealed IgA
deposition. Subsequent to intravenous glucocorticoid therapy and
prolonged maintenance treatment with prednisone, the blisters
disappeared without recurrence.
Pemphigus in the present patient appeared during
thalidomide maintenance therapy of the lymphoma. Thalidomide is
widely administered for the treatment of blood-lymphatic system
cancers due to its anti-angiogenesis effect and its dual
immunomodulatory function as an immunosuppressant or
immunostimulant (17). A study by
Herth et al (18) previously
concluded that thalidomide maintenance therapy drives the
maturation of T cells toward a memory phenotype, however,
compromises antigen-specific immunity. Additionally, the decreased
frequency and function of CD8+ T cells has been
previously described for T-cell lymphoma (19). It was hypothesized that the cause of
IgA pemphigus in the present patient may have been a combination of
the immunological imbalance, which was caused by the
blood-lymphatic system cancer, and thalidomide-induced
immunomodulatory effects. However, further evidence is required to
support this assumption.
There are no recognized guidelines for IgA pemphigus
management, however, the mainstay treatment of IgA pemphigus is
oral and topical corticosteroids. This treatment reduces
inflammation by reversing the increased permeability in capillaries
and terminating neutrophil activity (20). Dapsone and isotretinoin are also
effective treatments for IgA pemphigus (21,22).
Adalimumab and mycophenolate mofetil, which are effective in
treating classic pemphigus, have also been reported as proficient
treatments (23).
The present study is the first case of PTCL
complicated by IgA pemphigus in a Chinese patient. IgA-associated
autoimmune diseases, including IgA pemphigus, are clinically rare.
When a suspected case of IgA pemphigus is under investigation, an
immediate biopsy is recommended to acquire an early diagnosis. The
present case report of a patient with PTCL complicated with IgA
pemphigus facilitated the unique observation of the combination of
a malignant lymphoma with a particularly rare autoimmune disease.
Furthermore, the present case raises questions about the putative
effects of immunomodulatory maintenance therapy for individuals
that are autoimmune. In conclusion, the present study demonstrates
that investigation of the association between these two diseases is
necessary in future studies, as this association may determine the
phenomenon of neoplasia-induced autoimmunity.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 30900597) and the
Science Foundation of Hubei Health Department (grant no.
JX6B08).
References
|
1
|
Vose J, Armitage J and Weisenburger D;
International T-Cell Lymphoma Project. International peripheral
T-cell and natural killer/T-cell lymphoma study: pathology findings
and clinical outcomes. J Clin Oncol. 26:4124–4130. 2008.
|
|
2
|
Escalón MP, Liu NS, Yang Y, et al:
Prognostic factors and treatment of patients with T-cell
non-Hodgkin lymphoma: the M.D. Anderson Cancer Center experience.
Cancer. 103:2091–2098. 2005.
|
|
3
|
Martin DN, Mikhail IS and Landgren O:
Autoimmunity and hematologic malignancies: associations and
mechanisms. Leuk Lymphoma. 50:541–550. 2009.
|
|
4
|
Hashimoto T: Recent advances in the study
of the pathophysiology of pemphigus. Arch Dermatol Res. 295(Suppl
1): S2–S11. 2003.
|
|
5
|
Yeh SW, Ahmed B, Sami N and Razzaque Ahmed
A: Blistering disorders: diagnosis and treatment. Dermatol Ther.
16:214–223. 2003.
|
|
6
|
Hashimoto T: Immunopathology of IgA
pemphigus. Clin Dermatol. 19:683–689. 2001.
|
|
7
|
Hashimoto T, Kiyokawa C, Mori O, et al:
Human desmocollin 1 (Dsc1) is an autoantigen for the subcorneal
pustular dermatosis type of IgA pemphigus. J Invest Dermatol.
109:127–131. 1997.
|
|
8
|
Hashimoto T, Komai A, Futei Y, Nishikawa T
and Amagai M: Detection of IgA autoantibodies to desmogleins by an
enzyme-linked immunosorbent assay: the presence of new minor
subtypes of IgA pemphigus. Arch Dermatol. 137:735–738. 2001.
|
|
9
|
Amagai M, Komai A, Hashimoto T, et al:
Usefulness of enzyme-linked immunosorbent assay using recombinant
desmogleins 1 and 3 for serodiagnosis of pemphigus. Br J Dermatol.
140:351–357. 1999.
|
|
10
|
Ebihara T, Hashimoto T, Iwatsuki K, et al:
Autoantigens for IgA anti-intercellular antibodies of intercellular
IgA vesiculopustular dermatosis. J Invest Dermatol. 97:742–745.
1991.
|
|
11
|
Beutner EH, Chorzelski TP, Wilson RM, et
al: IgA pemphigus foliaceus. Report of two cases and a review of
the literature. J Am Acad Dermatol. 20:89–97. 1989.
|
|
12
|
Watanabe H, Okada M, Kaji Y, et al: New
response evaluation criteria in solid tumours-revised RECIST
guideline (version 1.1). Gan To Kagaku Ryoho. 36:2495–2501.
2009.(In Japanese).
|
|
13
|
Petropoulou H, Politis G, Panagakis P,
Hatziolou E, Aroni K and Kontochristopoulos G: Immunoglobulin A
pemphigus associated with immunoglobulin A gammopathy and lung
cancer. J Dermatol. 35:341–345. 2008.
|
|
14
|
Szturz P, Adam Z, Klincová M, et al:
Multiple myeloma associated IgA pemphigus: treatment with
bortezomib- and lenalidomide-based regimen. Clin Lymphoma Myeloma
Leuk. 11:517–520. 2011.
|
|
15
|
Taintor AR, Leiferman KM, Hashimoto T,
Ishii N, Zone JJ and Hull CM: A novel case of IgA paraneoplastic
pemphigus associated with chronic lymphocytic leukemia. J Am Acad
Dermatol. 56(5 Suppl): S73–S76. 2007.
|
|
16
|
Asahina A, Koga H, Suzuki Y and Hashimoto
T: IgA pemphigus associated with diffuse large B-cell lymphoma
showing unique reactivity with desmocollins: unusual clinical and
histopathological features. Br J Dermatol. 168:224–226. 2013.
|
|
17
|
Oxberry SG and Johnson MJ: Response to
thalidomide in chemotherapy-resistant cutaneous T-cell lymphoma.
Clin Oncol (R Coll Radiol). 18:86–87. 2006.
|
|
18
|
Herth I, Witzens-Harig M, Beckhove P, et
al: Thalidomide maintenance therapy maturates the T cell
compartment and compromises antigen-specific antitumor immunity in
patients with multiple myeloma. Exp Hematol. 41:231–240. 2013.
|
|
19
|
Kozako T: New treatment strategy for adult
T-cell leukemia targeting for anti-tumor immunity and a longevity
gene-encoded protein. Yakugaku Zasshi. 131:1061–1072. 2011.(In
Japanese).
|
|
20
|
Camisa C and Warner M: Treatment of
pemphigus. Dermatol Nurs. 10:115–118. 123–131. 1998.
|
|
21
|
Gruss C, Zillikens D, Hashimoto T, et al:
Rapid response of IgA pemphigus of subcorneal pustular dermatosis
type to treatment with isotretinoin. J Am Acad Dermatol.
43:923–926. 2000.
|
|
22
|
Ruiz-Genao DP, Hernández-Núñez A,
Hashimoto T, Amagai M, Fernández-Herrera J and García-Díez A: A
case of IgA pemphigus successfully treated with acitretin. Br J
Dermatol. 147:1040–1042. 2002.
|
|
23
|
Howell SM, Bessinger GT, Altman CE and
Belnap CM: Rapid response of IgA pemphigus of the subcorneal
pustular dermatosis subtype to treatment with adalimumab and
mycophenolate mofetil. J Am Acad Dermatol. 53:541–543. 2005.
|