Introduction
p130cas, also known as breast cancer anti-estrogen
resistance 1, is one of the Crk-associated substrate (cas) protein
family members. The designation Crk stands for CT10 regulator of
kinase, where CT10 is the avian virus from which a protein was
isolated, without kinase domains, but able to stimulate tyrosine
phosphorylation in cells (1).
p130cas was originally identified as a cellular protein migrating
at 130 kDa of molecular weight, and was hyperphosphorylated in
v-Crk- and v-Src-transformed cells (2). Our recent pilot studies indicated the
clinical and biological implications of p130cas in non-small cell
lung cancer (NSCLC) (3, 4). Serum p130cas levels were significantly
higher in NSCLC compared with the control group, gradually
increasing with the progression of tumor staging, and decreasing
following malignant lesion removal (3). In a cohort of 151 Chinese patients
with NSCLC, elevated p130cas protein expression levels in tumor
tissues were shown to predict a poor prognosis (hazard ratio,
1.777; P=0.028) (4). In addition,
p130cas-knockdown caused cell migration inhibition and arrest of
cell growth and the cell cycle in A549 lung cancer cells (4).
Elevating the expression of either the full-length
or just the carboxyl terminus of p130Cas in mammary epithelial
cells has been shown to diminish the ability of transforming growth
factor-β1 (TGF-β1) to activate Smad2/3, but increase its coupling
to p38 mitogen-activated protein kinases (MAPKs; p38) (4,5), whose
activation is required for TGF-β1 mediated fibroblastic
transdifferentiation and cell migration (6). Moreover, p38 inhibitors can block
TGF-β1-induced epithelial-mesenchymal transition (EMT) in a variety
of cells (6,7), including pulmonary epithelial cells
(7). The p38 signaling cascade has
significant roles in TGF-β1-induced metastasis, particularly in the
late stages of malignancy (8,9).
Our previous study demonstrated that
p130cas-knockdown reduced the phosphorylated-p38 (p-p38) level in
lung cancer A549 cells (4),
indicating the significance of p130cas in p38 activation and the
subsequent occurrence of EMT.
The present study evaluated the correlations between
p130cas-expression and the activation of p38 and Smad2, which are
components of the two main signaling pathways of TGF-β1, i.e., EMT
and apoptosis (10,11). The aim of the current study was to
clarify the critical roles of p130cas in the regulation of
TGF-β1-mediated EMT or apoptosis in A549 cells. Additionally, the
aim was to clarify the significant alteration of the TGF-β1
signaling pathways from tumor suppression to tumor promotion by the
regulation of p130cas.
Materials and methods
Cell culture and RNA interference (RNAi)
of p130cas in A549 cells
The A549 lung adenocarcinoma cell line was obtained
from the American Type Culture Collection (Manassas, VA, USA) and
cultured in RPMI 1640/10% fetal bovine serum.
The RNAi p130cas protocol was established according
to our previous published study (4). The following oligoribonucleotide pairs
were used: 5′-CCGGGG TCGACAGTGGTGTGTATTTCAAGAGAATACACACCAC
TGTCGACCTTTTTTg-3′ and 5′-AATTCAAAAAAGGTC
GACAGTGGTGTGTATTCTCTTGAAATACACACCACT GTCGACC-3′. Entire sequences
were derived from the sequence of human p130cas mRNA. The
oligonucleotides were obtained from Sunbio Medical Biotechnology
Co., Ltd. (Shanghai, China). The two complementary strands (each 20
mM) in 60 ml annealing buffer (Sunbio Medical Biotechnology Co.,
Ltd.) were heated for 5 min at 95°C and then incubated for 1 h at
room temperature. Thereafter, the green fluorescent protein
(GFP)-tagged lentiviral vector, pLVT351.LV for p130cas-RNAi, was
constructed by inserting the annealing nucleotides into the
AgeI+EcoRI site of pMAGic 4.1 (Sunbio Medical
Biotechnology Co., Ltd.).
Secondly, the A549 cell line was plated at
2.3×105 cells per well in a 24-well culture plate and
infected with lentivirus at a multiplicity of infection value of
10. The cells infected with pLVT351.LV and CMV-GFP-LV (blank
lentiviral vector; pMAGic 4.1) were termed the A549-p130cas-RNAi
and A549 negative control cells, respectively. The efficacy of the
RNAi of p130cas was confirmed by our previous study (4).
Quantitative reverse transcription
polymerase chain reaction (RT-qPCR)
In order to evaluate the efficacy of p130cas RNAi,
RT-qPCR was performed in A549-p130cas-RNAi and A549-negative
control cells. The cells were seeded at a concentration of
1×105 cells/well in 6-well plates. Two days after
seeding, total RNA was extracted from the cells using TRIzol
(Invitrogen Life Technologies, Carlsbad, CA, USA). First-strand
cDNA was synthesized with M-MLV transcriptase (Promega, Madison,
WI, USA) and oligo dT. RT-qPCR was performed using SYBR Green PCR
master mix (Takara Bio, Inc., Shiga, Japan) and the ABI Prism 7000
sequence detection system (Applied Biosystems, Foster City, CA,
USA). The following PCR primers were used: 5′-CAATGCCTCACTGCTCTT-3′
and 5′-GTAGTCATAGTCCTCCATC-3′. The specificity of detected signals
was confirmed by a dissociation curve consisting of a single peak.
All samples were performed in duplicate. Values were normalized by
human β-actin.
Cytokine treatment and cell morphology
inspection
The A549 cells were co-incubated at 37°C in a
humidified atmosphere of 5% CO2 with 7.5 ng/ml TGF-β1
(catalog no. 01-209; Upstate Biotechnology, New York, NY, USA) for
1, 12 and 36 h. Cell morphology was analyzed using light
microscopy, focusing on the changes of colonial morphology and
lamellipodia.
Cell invasiveness and migration
assay
For the assessment of cell motility, chamber
invasiveness and migration assays were conducted using a cell
culture insert (8-mm pore size, 24-well format; Cell Invasion Assay
kit, cat. no. ECM550; Chemicon, Temecula, CA, USA). The cells were
seeded in duplicate at a density of 3.0×105
cells/chamber. After 48 h, the cells which had not moved to the
lower wells were removed from the upper face of the filters using
cotton swabs, and the cells that had moved to the lower surface of
the filter were stained using a cell invasion assay kit, which
utilizes ECMatrix™, a reconstituted basement membrane matrix of
proteins derived from the Engelbreth Holm-Swarm mouse tumor. The
cells were quantified by visual counting once images had been
captured. Experiments were performed in triplicate. Mean values for
three random fields were obtained for each well.
Immunoblotting of biomarkers
A549 cell lysate was prepared by homogenization in a
RIPA buffer comprised of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1%
Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 0.5% deoxycholic
acid and 0.02% sodium azide. The protein concentrations were
determined with a Bicinchoninic Acid Protein Assay kit (Pierce,
Rockford, IL, USA). Proteins were denatured at 95°C for 5 min, and
50 mg protein per lane was resolved by SDS-polyacrylamide gel
electrophoresis using 10% polyacrylamide gel. Proteins were blotted
on polyvinylidene difluoride membranes (Thermo Fisher Scientific,
Waltham, MA, USA), which were then blocked with 5% skimmed milk for
1 h at room temperature. The proteins were immunoblotted using
anti-E-cadherin antibody (1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), anti-N-cadherin antibody (1:1,000; Cell
Signaling Technology, Inc.), anti-Vimentin antibody (1:200; Sigma
Aldrich, St. Louis, MO, USA), anti-Caspase-3 (Cleaved-Asp175)
antibody (1:1,000; Assay Biotechnology, Co., Inc., Sunnyvale, CA,
USA), anti-p-p38 antibody (Thr180/Tyr182; 1:1,000; Cell Signaling
Technology, Inc.), anti-p38 antibody (1:1,000; BD Transduction
Laboratories, BD Biosciences, Franklin Lakes, NJ, USA), anti-smad2
antibody (1:500; Abcam, Cambridge, MA, USA), anti-p-Smad2 antibody
(Ser467; 1:1,000; Bioworld Technology, St. Louis Park, MN, USA),
anti-p130cas antibody (1:1,000; BD Transduction Laboratories, BD
Biosciences) and anti-p-p130cas (Tyr165; 1:1,000; Cell Signaling
Technology, Inc.). An anti-GAPDH (Sigma Aldrich) antibody served as
the control. Experiments were performed in triplicate.
Immunoblotting was quantified with Quantity One software (Bio-Rad,
Hercules, CA, USA). The analyses of the bands of the different
proteins were referenced against GAPDH.
Data analysis
The statistical analysis of the association between
the protein levels was performed using Pearson’s correlation
analysis. Student’s t-test was used to evaluate differences in the
variables (mean ± standard error of the mean) between the two study
groups. The analysis was performed using SPSS Version 11.0 software
for Windows (SPSS, Inc., Chicago, IL, USA). P<0.05 (two-sided)
was considered to indicate a statistically significant
difference.
Results
p130cas-knockdown reduces p-p38 levels in
A549 cells and blockades phosphorylation of p38 induced by TGF-β1,
but has no impact on the activation of Smad2
A549 cells that stably expressed pLVT351-LV
(A549-p130cas-RNAi cells) and CMVGFP-LV (A549-negative control
cells) were established through use of a lentivirus system
(4). At least an 80% reduction in
p130cas-mRNA and -protein in A549-p130cas-RNAi cells was confirmed
by RT-qPCR (4), and western
blotting analysis (Fig. 1A),
respectively. Additionally, the expression level of p-p130cas was
reduced markedly following p130cas-RNAi (Fig. 1A). TGF-β1 treatment markedly
increased the expression level of p130cas in the A549-negative
control cells and the A549-p130cas RNAi cells (Fig. 1A).
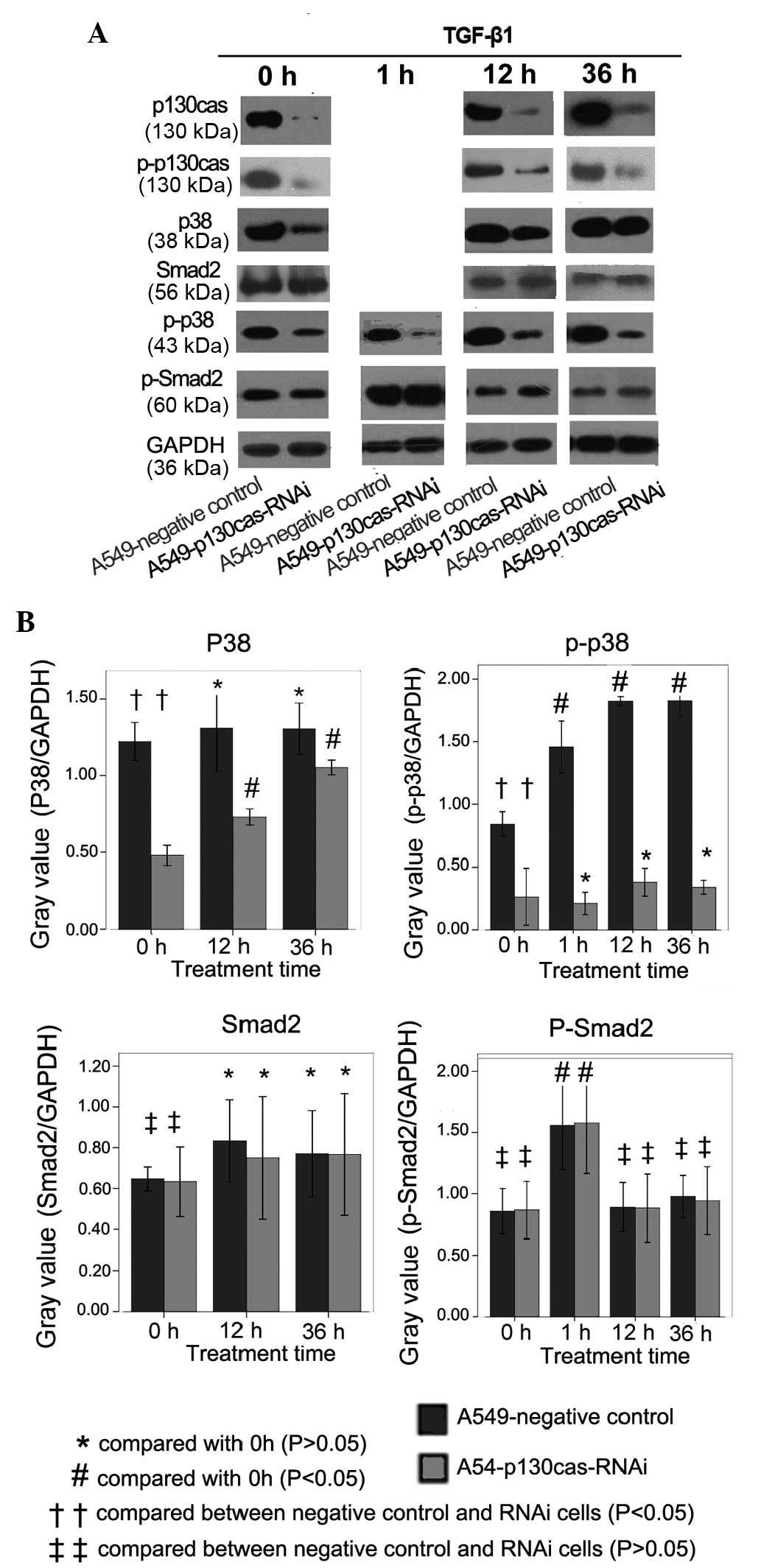 | Figure 1(A) Immunoblotting was conducted to
detect the expression levels of p130cas, p-p130cas, p38, p-p38,
Smad2 and p-Smad2. The expression levels of p130cas-protein and
p-p130cas were reduced markedly in the A549-p130cas-RNAi cells
compared with the negative control cells. TGF-β1 treatment (7.5
ng/ml) for 12 h or 36 h markedly increased the p130cas expression
levels in the A549 cells. The expression levels of total-p38 and
p-p38 were reduced significantly in the A549-p130cas-RNAi cells
compared with the A549-negative control cells. The total-p38
expression level was significantly upregulated in the
A549-p130cas-RNAi cells following TGF-β1 treatment, however, the
p-p38 expression level in the A549-p130cas-RNAi cells was
consistently significantly lower than in the A549-negative control
cells prior to or following TGF-β1 treatment. TGF-β1 remarkably
increased the expression levels of p-Smad2 in the A549 cells
following 1 h of treatment. However, p130cas-knockdown had no
impact on the expression of total-Smad2 or p-Smad2 compared with
the control cells. (B) Quantification of immunoblotting was
conducted with Quantity One software. The analyses of the bands of
the different proteins were reference against GAPDH. Mean ± SEM
values were calculated in order to establish any statistically
significant differences. TGF-β1, transforming growth factor-β1;
RNAi, RNA interference; cas, Crk-associated substrate. |
The expression levels of total-p38 and p-p38 were
reduced significantly in the A549-p130cas-RNAi cells compared with
the A549-negative control cells (P=0.02 and 0.01, respectively;
Fig. 1A and B). Notably, the
total-p38 expression level was significantly upregulated in the
A549-p130cas-RNAi cells following TGF-β1 treatment (P=0.02;
Fig. 1A and B), however, the p-p38
expression level in the A549-p130cas-RNAi cells was consistently
significantly (P=0.01; Fig 1A and
B) lower than in the A549-negative control cells prior to and
following TGF-β1 treatment (P=0.008; Fig. 1A and B), indicating that the
TGF-β1-induced phosphorylation of p38 was effectively blocked by
p130cas-knockdown.
In addition, TGF-β1 markedly increased the
expression levels of p-Smad2 in the A549-negative control cells and
the A549-p130cas-RNAi cells following 1 h of treatment (Fig. 1A and B). However, p130cas-knockdown
had no impact on the expression of total-Smad2 or p-Smad2 compared
with the control cells (Fig. 1A and
B).
p130cas-knockdown arrests cell invasion
and the EMT induced by TGF-β1 in A549 cells, but has no impact on
the cleavage of Capase-3
Co-incubation of TGF-β1 and epithelial cells is a
convenient way to induce EMT (11,12),
which may be characterized by changes in cell shape, cell migration
and biomarkers, e.g., the downregulation of E-Cadherin and the
upregulation of N-Cadherin (13).
Therefore, the following investigations were conducted in the
present study in order to clarify the impact of p130cas on
TGF-β1-induced EMT in A549 cells.
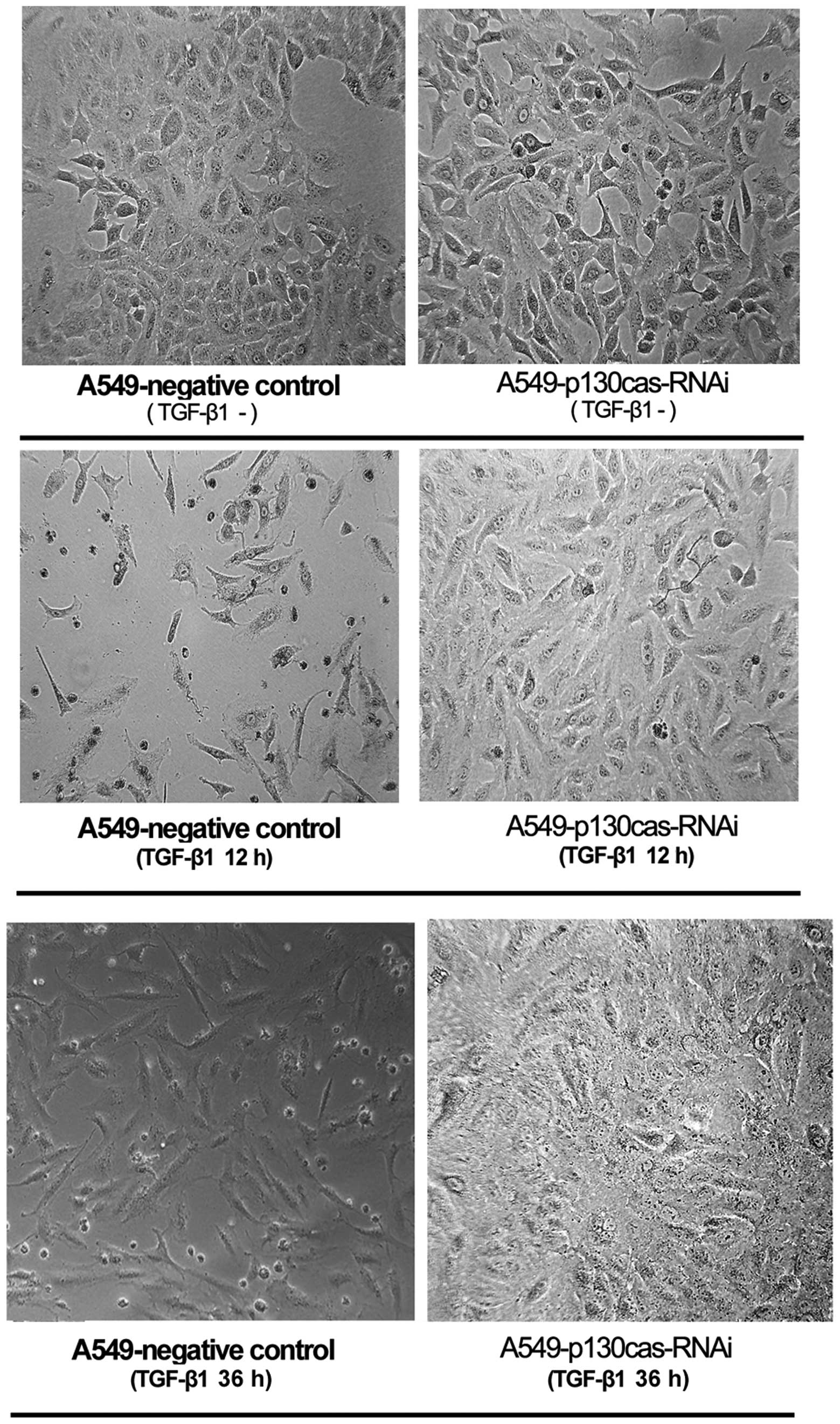
In the A549-negative control cells, TGF-β1 treatment
(12 h or 36 h) induced EMT-associated cell shapes, including loss
of colonial morphology and increased lamellipodia (Fig. 2). However, TGF-β1 treatment did not
induce these morphological changes in the A549-p130cas-RNAi cells
(Fig. 2).
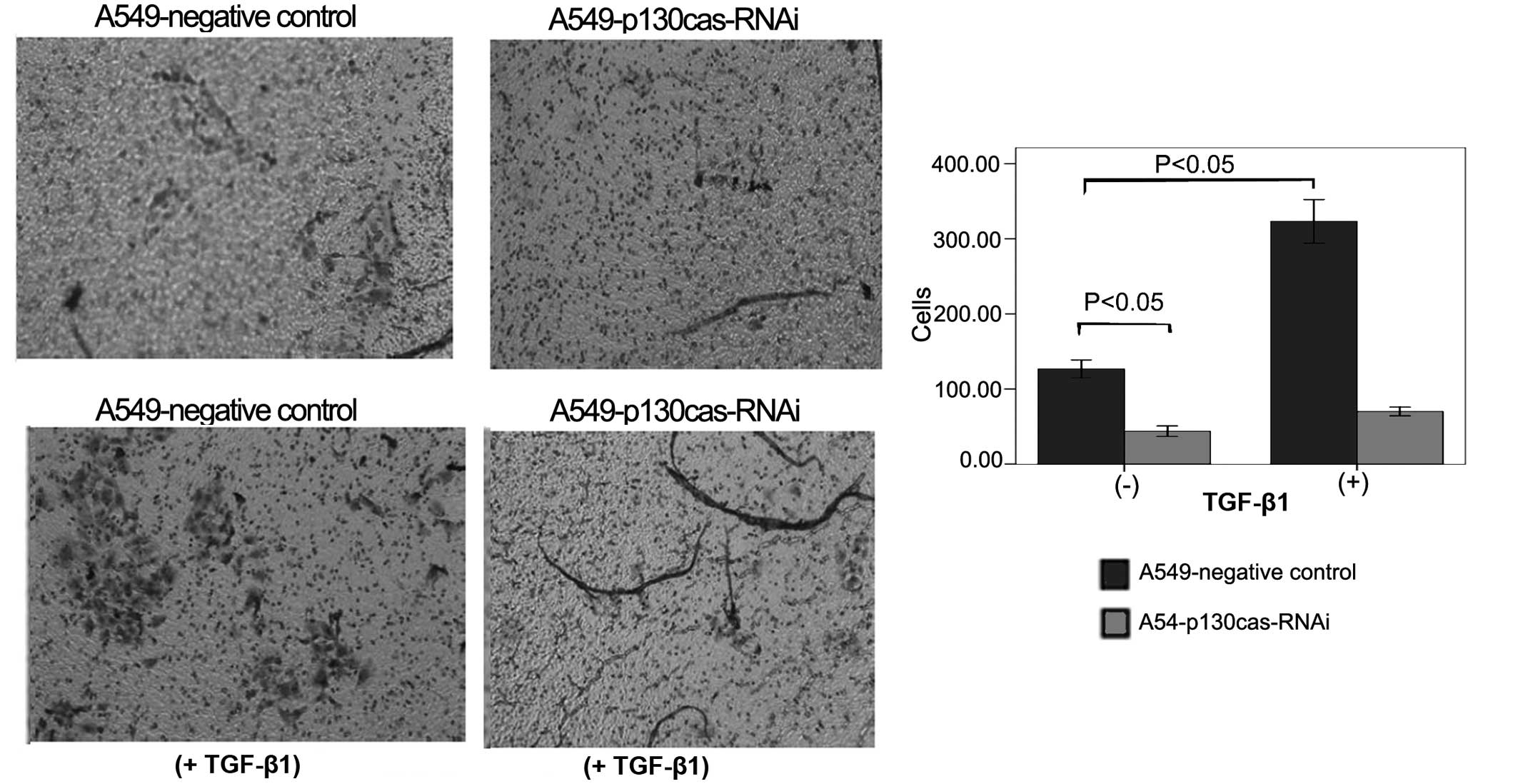
Subsequent to 48 h of treatment, TGF-β1 markedly
stimulated the migration of the A549-negative control cells (130±10
vs. 320±25, P=0.0003; Fig. 3) and
the A549-p130cas-RNAi cells (43±6 vs. 70±6, P=0.005; Fig. 3), respectively. However, the
migrated cells of the p130cas-RNAi group were consistently
significantly less in number compared with those of the negative
control group prior to or following TGF-β1 treatment (43±6 vs.
130±10, P=0.0002; 70±6 vs. 320±25, P=0.0001; Fig. 3).
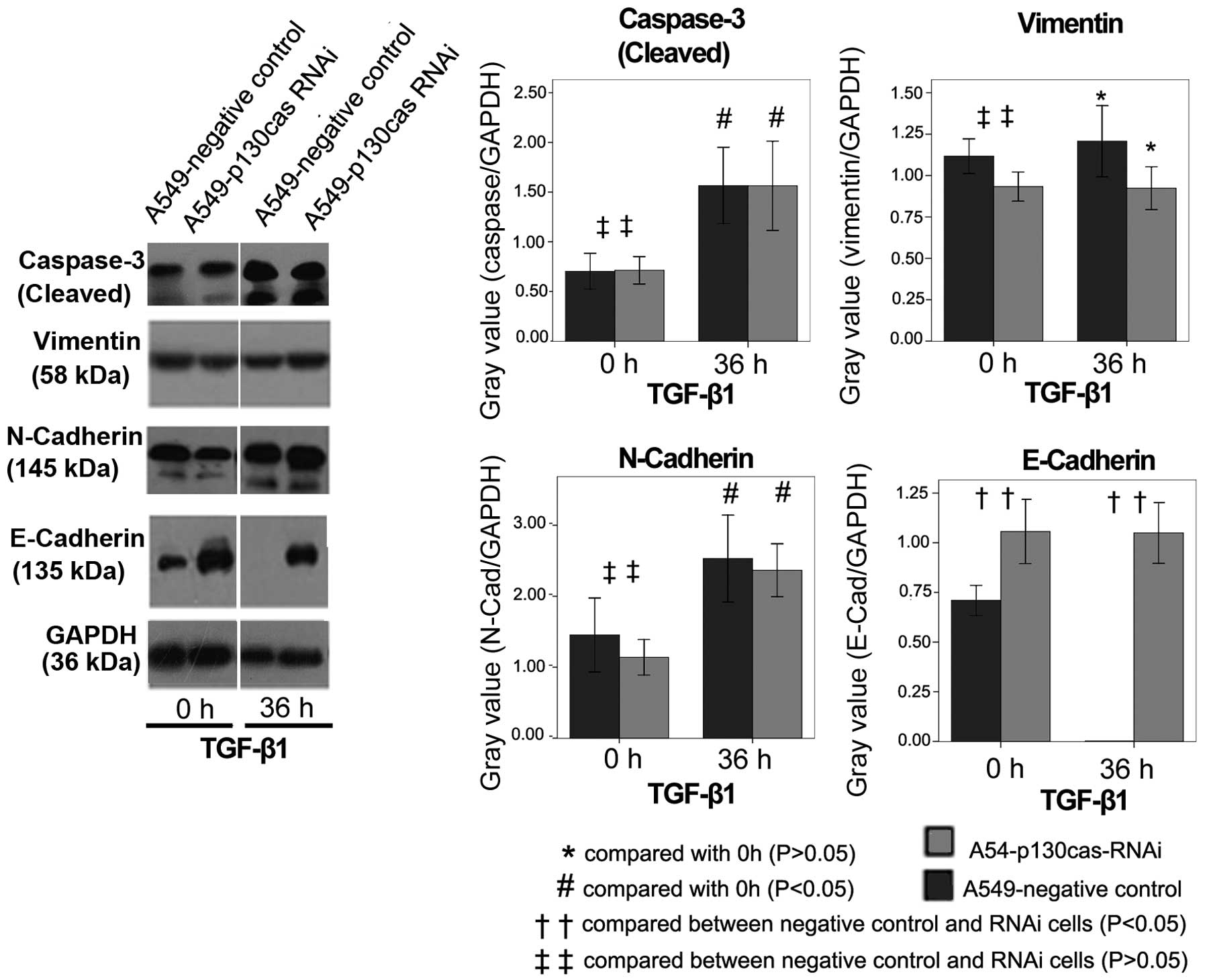
E-Cadherin expression levels were significantly
higher in A549-p130cas-RNAi cells than in A549-negative control
cells (P=0.0001; Fig. 4). After 36
h incubation with TGF-β1, E-Cadherin was consistently abundant in
A549-p130cas-RNAi cells, but undetectable in A549-negative control
cells (Fig. 4). N-cadherin levels
were slightly lower in A549-p130cas-RNAi cells than in
A549-negative control cells (Figure
4). TGF-β1 remarkably increased N-Cadherin in either
A549-p130cas-RNAi or negative control cells after 36 h treatment
(Fig. 4). There was no evident
disparity in Vimentin levels prior to or following TGF-β1 treatment
(Fig. 4).
Cleaved Caspase-3 levels were markedly increased in
the A549-p130cas-RNAi cells and negative control cells following 36
h of TGF-β1 treatment (Fig. 4).
Significantly, p130cas-knockdown had no impact on the cleavage of
Caspase-3 (P=0.82; Fig. 4) compared
with the control cells.
Discussion
A number of studies (14–16)
have demonstrated evidence of EMT, which is a critical event in
cancer progression, characterized by morphological changes,
enhanced cell motility, downregulation of epithelial markers and
up-regulation of mesenchymal markers (13), in a variety of malignancies. TGF-β1
has been found to be upregulated in a variety of tumors (17–19),
and plays significant roles in EMT and metastasis (12). Co-incubation of TGF-β1 and
epithelial cells is a convenient way to induce EMT in various cells
(12). Thus far, the p38 pathway,
also known as the non-canonical TGF-β1 signaling pathway (20), has been found to be critical in
TGF-β1-mediated EMT (7), and p38
inhibitors have been shown to block TGF-β1-induced EMT in a variety
of cell lines (6). Using
immunoblotting, Greenberg et al (21) found that compared with normal
tissue, only activated p38 MAPK was consistently increased in
NSCLC, indicating that this pathway has additional roles in
malignant cell growth or transformation. In our previous study,
p130cas was shown to be critical in the activation of p38 in
vivo and in vitro (4).
To the best of our knowledge, few studies have focused on the
issues analyzed in the present study of whether there is any
association between p130cas expression and TGF-β1-induced EMT in
lung cancer, and whether p130cas-knockdown is able to inhibit
TGF-β1-induced EMT in lung cancer.
A549 lung cancer is a well-characterized cell line
that has been used as a model system to study the underlying
mechanisms of carcinogenesis, apoptosis and cancer progression in
lung cancer (22). Moreover, A549
cells are conventionally used to study EMT (22). Hence, it was chosen as a model
system for the present study.
In the A549-negative control cells, TGF-β1 treatment
led to the marked elimination of E-Cadherin, which is the prototype
epithelial cell marker of EMT, whose expression has been found to
be downregulated markedly during EMT (23). Additionally, TGF-β1 treatment led to
the increased expression of N-Cadherin, which is a mesenchymal
marker (24), indicating the
occurrence of EMT in the A549-negative control cells. However,
TGF-β1 treatment did not result in any elimination of E-Cadherin
expression in the A549-p130cas-RNAi cells. Meanwhile, the
expression levels of N-Cadherin were slightly lower in the
p130cas-RNAi cells compared with the control cells. All the results
indicated that p130cas-knockdown resulted in the inhibition of EMT,
leading to changes in the expression of epithelial and mesenchymal
markers. Recently, Miao et al (25) also found that the overexpression of
p130cas was significantly correlated with the decreased expression
of E-cadherin in 105 NSCLC tissues (P=0.002). Although the study by
Miao et al (in vivo) and the present study (in
vitro) simultaneously supported that the significantly adverse
association between the expression of p130cas and E-cadherin in
NSCLC, the underlying mechanism remains unknown. Tikhmyanova and
Golemis (26) found that cas
proteins promoted the lysosomal degradation of E-cadherin via Src
kinase in MCF7 breast adenocarcinoma cells. We hypothesize that
there is similar mechanism for the p130cas-induced lysosomal
degradation of E-cadherin in NSCLC, which warrants further
study.
In the present study, the cell shapes of the
A549-negative control cells, characterized by the loss of colonial
morphology and increased lamellipodia, following TGF-β1 treatment
strongly demonstrated the occurrence of EMT. However, TGF-β1 did
not stimulate EMT-associated morphological changes in the
p130cas-RNAi cells. Moreover, Transwell cell assays supported the
fact that p130cas-knockdown significantly blocked the enhancement
of cellular motility induced by TGF-β1.
Therefore, p130cas has been shown to be required for
TGF-β1-mediated EMT in A549 cells. However, the underlying
mechanisms warrant further investigations. The present study
indicated that p130cas-knockdown decreased the expression level of
p-p38 in the A549 cells and blockaded the TGF-β1-induced activation
of p-p38. The p38 MAPK signaling pathway has been found to be
involved in the phosphorylation of Twist1, the stabilization of
Twist1 protein and the induction of EMT in mammary epithelial cells
(27). In addition, the close
correlations between Snail (an E-Cadherin repressor) and
phospho-p38 expression levels have been confirmed in ovarian
carcinoma, demonstrating the ability of p38 MAPK to induce EMT via
Snail (28). Moreover, p38 MAPK is
required in TGF-β1-induced EMT via fibronectin upregulation and
collagen expression (7). The
published literature therefore strongly supports our hypothesis
regarding the requirement of p130cas for TGF-β1-mediated EMT in
NSCLC, potentially due to the activation of p38 MAPK.
In addition to p38 activation, TGF-β1 treatment has
been shown to upregulate the expression of p-smad2 (29), which is traditionally believed to be
an inducer of apoptosis and a tumor suppressor (10,11,30).
The present study showed that the levels of p-smad2 peaked
following 1 h of treatment with TGF-β1, and then decreased to
almost the same level as the controls after 12 h, concordant with
the results of other published studies (29–33).
The present study indicated that p130cas-knockdown had no marked
impact on the activation of Smad2 with or without TGF-β1 treatment.
However, in breast cancer cells, p130cas overexpression has been
shown to decrease the ability of TGF-β1 to activate Smad2/3, while
depleting p130cas led to the increased activation of Smad2/3
(5). We postulate that the mutants
of Smad2 in lung cancer A549 cells (34) led to the loss of cross-talk between
p130cas and Smad2-activation.
In the present study, p130cas-knockdown had no
impact on the expression of cleaved Caspase-3, which is a marker of
apoptosis. Our previous study also indicated that the rate of
apoptosis following either transient (39.1 vs. 42.1%) or stable
transfection (0.33 vs. 0.4%) in A549 cells was not increased by
p130cas-knockdown, as observed by FACScan flow cytometry assays
(4). All the aforementioned results
have indicated that p130cas has no evident association with the
apoptosis of A549 cells.
Notably, the p38 MAPK pathway can also induce
apoptosis, and inhibitors of p38 MAPK induce anoikis resistance in
human breast cancer cells and murine mammary epithelial cells
(35,36). The p38 MAPK pathway induces
apoptosis and anoikis most likely due to the activation of
mitochondrial Bax and subsequent cytochrome c release
(37). Determining the mechanism by
which p130cas blocks p38-rendered apoptosis will be the aim of our
future study.
In conclusion, the overexpression of p130cas has
significant associations with p-p38 activation in vivo and
in vitro. The present study showed that p130cas had a
critical role in TGF-β1-induced EMT in the A549 cells, but no
impact on TGF-β1-induced apoptosis. p130cas is a novel molecular
‘rheostat’ that alters the function of the TGF-β1 signaling pathway
from tumor suppression to tumor promotion in lung cancer cells.
Acknowledgements
The authors would like to thank Professor Li Zeng
Peng and his staff from the Department of Pathology at Daping
Hospital (Chongqing, China) for conducting immunohistochemistry
tests and reviewing slides. In addition, the authors would like to
thank the academic editor and anonymous reviewers for their
comments on the manuscript. The present study was supported by
grants from the National Natural Science Foundation of China (NSFC;
no. 81101782), the NSF project CQ CSTC (no. CSTC2011BB5020) and the
NSF Third Military Medical University (no. 2010XQN36 and MiaoPu
project).
References
|
1
|
Mayer BJ, Hamaguchi M and Hanafusa H: A
novel viral oncogene with structural similarity to phospholipase C.
Nature. 332:272–275. 1988.
|
|
2
|
Kanner SB, Reynolds AB and Parsons JT:
Tyrosine phosphorylation of a 120-kilodalton pp60src substrate upon
epidermal growth factor and platelet-derived growth factor receptor
stimulation and in polyomavirus middle-T-antigen-transformed cells.
Mol Cell Biol. 11:713–720. 1991.
|
|
3
|
Deng B, Huang W, Tan QY, et al: Breast
cancer anti-estrogen resistance protein 1 (BCAR1/p130cas) in
pulmonary disease tissue and serum. Mol Diagn Ther. 15:31–40.
2011.
|
|
4
|
Huang W, Deng B, Wang RW, et al: BCAR1
protein plays important roles in carcinogenesis and predicts poor
prognosis in non-small-cell lung cancer. PLoS One.
7:e361242012.
|
|
5
|
Wendt MK, Smith JA and Schiemann WP:
p130Cas is required for mammary tumor growth and transforming
growth factor-beta-mediated metastasis through regulation of
Smad2/3 activity. J Biol Chem. 284:34145–34156. 2009.
|
|
6
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFbeta-mediated fibroblastic transdifferentiation and cell
migration. J Cell Sci. 115:3193–3206. 2002.
|
|
7
|
Kolosova I, Nethery D and Kern JA: Role of
Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial-mesenchymal
transition of pulmonary epithelial cells. J Cell Physiol.
226:1248–1254. 2011.
|
|
8
|
Galliher AJ, Neil JR and Schiemann WP:
Role of transforming growth factor-beta in cancer progression.
Future Oncol. 2:743–763. 2006.
|
|
9
|
del Barco Barrantes I and Nebreda AR:
Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans.
40:79–84. 2012.
|
|
10
|
Yang J, Song K, Krebs TL, Jackson MW and
Danielpour D: Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced
apoptosis and tumor progression. Oncogene. 27:5326–5338. 2008.
|
|
11
|
Yang J, Wahdan-Alaswad R and Danielpour D:
Critical role of Smad2 in tumor suppression and transforming growth
factor-beta-induced apoptosis of prostate epithelial cells. Cancer
Res. 69:2185–2190. 2009.
|
|
12
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009.
|
|
13
|
Nelson CM, Khauv D, Bissell MJ and Radisky
DC: Change in cell shape is required for matrix
metalloproteinase-induced epithelial-mesenchymal transition of
mammary epithelial cells. J Cell Biochem. 105:25–33. 2008.
|
|
14
|
Hayashida T, Jinno H, Kitagawa Y and
Kitajima M: Cooperation of cancer stem cell properties and
epithelial-mesenchymal transition in the establishment of breast
cancer metastasis. J Oncol. 2011:5914272011.
|
|
15
|
Lo JF, Yu CC, Chiou SH, et al: The
epithelial-mesenchymal transition mediator S100A4 maintains
cancer-initiating cells in head and neck cancers. Cancer Res.
71:1912–1923. 2011.
|
|
16
|
Maitah MY, Ali S, Ahmad A, Gadgeel S and
Sarkar FH: Up-regulation of sonic hedgehog contributes to
TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells.
PLoS One. 6:e160682011.
|
|
17
|
Joshi A and Cao D: TGF-beta signaling,
tumor microenvironment and tumor progression: the butterfly effect.
Front Biosci (Landmark Ed). 15:180–194. 2010.
|
|
18
|
Yang L, Pang Y and Moses HL: TGF-beta and
immune cells: an important regulatory axis in the tumor
microenvironment and progression. Trends Immunol. 31:220–227.
2010.
|
|
19
|
Taylor MA, Lee YH and Schiemann WP: Role
of TGF-β and the tumor microenvironment during mammary
tumorigenesis. Gene Expr. 15:117–132. 2011.
|
|
20
|
Iwata J, Hacia JG, Suzuki A, Sanchez-Lara
PA, Urata M and Chai Y: Modulation of noncanonical TGF-β signaling
prevents cleft palate in Tgfbr2 mutant mice. J Clin Invest.
122:873–885. 2012.
|
|
21
|
Greenberg AK, Basu S, Hu J, et al:
Selective p38 activation in human non-small cell lung cancer. Am J
Respir Cell Mol Biol. 26:558–564. 2002.
|
|
22
|
Pirozzi G, Tirino V, Camerlingo R, et al:
Epithelial to mesenchymal transition by TGFβ-1 induction increases
stemness characteristics in primary non small cell lung cancer cell
line. PLoS One. 6:e215482011.
|
|
23
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005.
|
|
24
|
Araki K, Shimura T, Suzuki H, et al:
E/N-cadherin switch mediates cancer progression via TGF-β-induced
epithelial-to-mesenchymal transition in extrahepatic
cholangiocarcinoma. Br J Cancer. 105:1885–1893. 2011.
|
|
25
|
Miao Y, Li AL, Wang L, et al: Expression
of p130cas, E-cadherin and β-catenin and their correlation with
clinicopathological parameters in non-small cell lung cancer:
p130cas over-expression predicts poor prognosis. Folia Histochem
Cytobiol. 50:392–397. 2012.
|
|
26
|
Tikhmyanova N and Golemis EA: NEDD9 and
BCAR1 negatively regulate E-cadherin membrane localization, and
promote E-cadherin degradation. PLoS One. 6:e221022011.
|
|
27
|
Hong J, Zhou J, Fu J, et al:
Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1
protein and promotes breast cancer cell invasiveness. Cancer Res.
71:3980–3990. 2011.
|
|
28
|
Hipp S, Berg D, Ergin B, et al:
Interaction of Snail and p38 mitogen-activated protein kinase
results in shorter overall survival of ovarian cancer patients.
Virchows Arch. 457:705–713. 2010.
|
|
29
|
Ramos C, Becerril C, Montaño M, et al:
FGF-1 reverts epithelial-mesenchymal transition induced by
TGF-{beta}1 through MAPK/ERK kinase pathway. Am J Physiol Lung Cell
Mol Physiol. 299:L222–L231. 2010.
|
|
30
|
Miles FL, Tung NS, Aguiar AA, Kurtoglu S
and Sikes RA: Increased TGF-β1-mediated suppression of growth and
motility in castrate-resistant prostate cancer cells is consistent
with Smad2/3 signaling. Prostate. 72:1339–1350. 2012.
|
|
31
|
Kasai H, Allen JT, Mason RM, Kamimura T
and Zhang Z: TGF-beta1 induces human alveolar epithelial to
mesenchymal cell transition (EMT). Respir Res. 6:562005.
|
|
32
|
Liu LC, Tsao TC, Hsu SR, et al: EGCG
inhibits transforming growth factor-β-mediated
epithelial-to-mesenchymal transition via the inhibition of Smad2
and Erk1/2 signaling pathways in nonsmall cell lung cancer cells. J
Agric Food Chem. 60:9863–9873. 2012.
|
|
33
|
Filyak Y, Filyak O, Souchelnytskyi S and
Stoika R: Doxorubicin inhibits TGF-beta signaling in human lung
carcinoma A549 cells. Eur J Pharmacol. 590:67–73. 2008.
|
|
34
|
Yanagisawa K, Uchida K, Nagatake M, et al:
Heterogeneities in the biological and biochemical functions of
Smad2 and Smad4 mutants naturally occurring in human lung cancers.
Oncogene. 19:2305–2311. 2000.
|
|
35
|
Lagadec C, Meignan S, Adriaenssens E, et
al: TrkA overexpression enhances growth and metastasis of breast
cancer cells. Oncogene. 28:1960–1970. 2009.
|
|
36
|
Zhang Y, Rivera Rosado LA, Moon SY and
Zhang B: Silencing of D4-GDI inhibits growth and invasive behavior
in MDA-MB-231 cells by activation of Rac-dependent p38 and JNK
signaling. J Biol Chem. 284:12956–12965. 2009.
|
|
37
|
Owens TW, Valentijn AJ, Upton JP, et al:
Apoptosis commitment and activation of mitochondrial Bax during
anoikis is regulated by p38MAPK. Cell Death Differ. 16:1551–1562.
2009.
|