Introduction
The regulation of gene expression is contributed to
by epigenetic mechanisms that affect the chromatin structure.
Aberrant DNA methylation has been found in almost all tumor types,
including leukemia (1–3), and is associated with the aberrant
silencing of tumor suppressor genes in cancer. However, the
demethylation of the promoter-associated CpG islands leads to gene
re-expression. 5-Aza-2′-deoxycytidine (also known as decitabine;
DAC) is a cytosine nucleoside analogue that shows noteworthy
antineoplastic activity in patients with myelodysplastic syndromes
(4–6). The antitumor activity of DAC is
mediated by the inhibition of DNA methylation, which results in the
activation of specific genes. DAC has also been demonstrated to
activate the expression of tumor suppressor genes, including
p16INK4a (p16) and retinoic acid receptor β (RAR-β), which are
silenced by DNA methylation (7–9).
All-trans retinoic acid (ATRA) is a potent inducer of the
granulocytic differentiation of myeloid leukemia cells (10,11)
and has been used successfully to treat acute promyelocytic
leukemia. However, the antineoplastic effect of the two drugs in
combination on acute erythroleukemia has not previously been
reported. The current study investigated the antineoplastic
activity of DAC and ATRA in the human erythroleukemia K562 cell
line in vitro, as well as the effects of these agents on the
tumor suppressor genes, p16 and RAR-β.
Materials and methods
Materials
DAC and ATRA were purchased from Sigma-Aldrich (St.
Louis, MO, USA). DAC was dissolved in 0.45% NaCl containing 10 mM
sodium phosphate (pH 6.8) and stored at −80°C, while ATRA was
dissolved in absolute ethanol, protected from light and stored at
−20°C. The concentration of the DAC and ATRA stock solutions was
100 μmol/l.
Cells lines, cell culture and drug
treatments
The human erythroleukemia K562 cell line was
provided by the Jiangsu Institute of Hematology (Suzhou, China).
The cells were cultured in suspension in Roswell Park Memorial
Institute-1640 medium (Gibco, Grand Island, NY, USA) supplemented
with 10% fetal bovine serum (Gibco) and incubated in standard
tissue culture incubators at 37°C in a humidified atmosphere
containing 5% CO2. Various concentrations of DAC and
ATRA, alone or in combination, were added to the medium
simultaneously. Briefly, the cells were treated with drugs as a
sequential exposure for 24, 48 and 72 h using the following
experimental conditions: 1 μmol/l DAC, 2 μmol/l DAC, 4 μmol/l DAC,
0.5 μmol/l ATRA, 1 μmol/l DAC plus 0.5 μmol/l ATRA, 2 μmol/l DAC
plus 0.5 μmol/l ATRA and 4 μmol/l DAC plus 0.5 μmol/l ATRA. The
untreated K562 cells were used as a control.
Cell growth inhibition assay
Cell growth inhibition activity was determined using
a water-soluble tetrazolium-1 (WST-1) cell proliferation and
cytotoxicity assay kit (Beyotime, Haimen, China), according to the
manufacturer’s instructions. Briefly, 100 μl cell suspension
(containing 5×104 cells) was plated in each well of
96-well plates. The cells were then treated with the two drugs as
aforementioned for 24, 48 and 72 h. At the end of each experiment,
the cell proliferation reagent, WST-1 (10 μl), was added to each
well and the cells were incubated at 37°C for 4 h. The absorbance
(A) at 450 nm of each well was measured using a spectrophotometer
and the inhibitory rate was calculated using the following formula:
Inhibitory rate (%) = [1 - (Adrug - Ablank) /
(Acontrol - Ablank)] × 100. Three independent
experiments were performed in quadruplicate.
Cell differentiation and apoptosis
assay
The expression of the myelomonocytic antigens,
cluster of differentiation (CD)11b, CD14, CD13 and CD33, on the
cell surface was determined by direct immunofluorescence staining
and flow cytometry. Briefly, the cells were collected and washed
with phosphate-buffered saline (Beyotime). A total of
5×105 cells were stained with the following conjugated
antibodies: CD11b-phycoerythrin (PE; catalogue number: 555388),
CD14-fluorescein isothiocyanate (FITC; catalogue number: 555397),
CD13-PE and CD33-FITC (catalogue numbers 555394 and 555626,
respectively, BD Biosciences, Franklin Lakes, NJ, USA). The cells
were incubated for 15 min at 4°C and then analyzed using a flow
cytometer (FACScabilur; BD Biosciences). Apoptosis assays were
performed using an Annexin V-FITC apoptosis detection kit
(Beyotime) according the manufacturer’s instructions, and early
apoptosis was evaluated by cytofluorometry (FACScabilur, BD
Biosciences).
Methylation-specific polymerase chain
reaction (MSP)
MSP was used to determine the methylation status at
the 5′CpG island in the p16 and RAR-β promoter regions. Bisulfite
converts unmethylated cytosine residues to uracil, but methylated
cytosines remain non-reactive. PCR amplifies uracil as thymine,
while methylated cytosines are only amplified as cytosines. MSP
distinguishes unmethylated from methylated alleles in a given gene
based on sequence changes following bisulfite treatment of the DNA
using primers designed for methylated or unmethylated DNA. The
cells of different groups were collected for MSP at 48 h
post-incubation. The DNA from the cell line was extracted using the
ZR Genomic DNA II kit (Zymo Research Corporation, Irvine, CA, USA)
as recommended by the manufacturer. Bisulfite modification of the
genomic DNA was performed using the EZ DNA Methylation-Gold kit
(Zymo Research Corporation) according to the manufacturer’s
instructions. The PCR amplification was performed using p16 and
RAR-β promoter gene fragment-specific primers for methylated or
unmethylated DNA (Sangon Biotech Co., Ltd., Shanghai, China). The
primers used for unmethylated p16 were: Sense, 5′-TTTTTGGTG
TTAAAGGGTGGTGTACT-3′ and antisense, 5′-CACAAA
AACCCTCACTCACAACAA-3′, which yielded a fragment of 132 bp. The
primers used for methylated p16 were: Sense,
5′-GTGTTAAAGGGCGGCGTAGC-3′ and antisense, 5′-AAA
ACCCTCACTCGCGACGA-3′, which yielded a PCR product of 122 bp. The
primers used for unmethylated RAR-β were: Sense,
5′-TGGGATGTTGAGAATGTGAGTGATTT-3′ and antisense,
5′-CTTACTCAACCAATCCAACCAAAACA-3′, which yielded a fragment of 160
bp. The primers used for methylated RAR-β were: Sense,
5′-GGATTGGGATGTCGAGAACGC-3′ and antisense, 5′-CGACCAATCCAACCGAAACG
-3′, which yielded a PCR product of 158 bp. The PCR was performed
under the following conditions: 95°C for 4 min; 94°C for 25 sec,
62°C (p16) or 64°C (RAR-β) for 25 sec and 72°C for 30 sec for 25
cycles; and 72°C for 5 min. The CpGenome universal methylated DNA
(Millipore Corporation, Billerica, MA, USA) was used as a control
for the methylated DNA. The PCR-amplified products were separated
by electrophoresis on 2% agarose gel and visualized by ethidium
bromide staining under ultraviolet light. Images were then
captured.
RNA extraction and cDNA conversion
Following incubation with DAC and ATRA for 48 h, the
cells were harvested for RNA extraction. Total RNA was extracted
from freshly isolated culture cells, using the TRIzol one-step
procedure (Invitrogen Life Technologies, Paisley, United Kingdom),
according to the manufacturer’s instructions, and dissolved in
diethylpyrocarbonate-treated water. Reverse transcription was
performed using random hexamer primers for total RNA (2 μg/40 μl),
and 100 units of MuLV reverse transcriptase (Fermentas, Hanover,
MD, USA) was added to the reaction mixture, obtaining a significant
enhancement of the assay sensitivity. The cDNA was stored at
−20°C.
Quantitative PCR (qPCR)
qPCR was performed using the 7500 Fast Real-Time PCR
system (Applied Biosystems, Foster City, CA, USA) and all the
primers were synthesized by Sangon Biotech Co., Ltd. The primers
and probes specific for the p16 and RAR-β genes were as follows:
p16 sense, 5′-CTGCCCGTGGACCTGGC-3′ and antisense, 5′-CTC
TGGTTCTTTCAATCGGGG-3′; p16 TaqMan probe, 5′-AGT
AACCATGCCCGCATAGATGCCG-3′; RAR-β sense,
5′-AGATCGTGGAGTTTGCTAAACGT-3′ and antisense,
5′-GGGTATACCTGGTGCAAATTCTAAG-3′; and RAR-β TaqMan probe,
5′-CAAATTACCCTGCTGAAGGCCGCC-3′. GAPDH was utilized as the
housekeeping gene as an internal control of the RNA quality. The
primers and probes used were as follows: GADPH sense,
5′-GGAAGGTGAAGGTCGGAGTC-3′ and antisense, 5′-CGT
TCTCAGCCTTGACGGT-3′; and GADPH TaqMan probe,
5′-TTTGGTCGTATTGGGCGCCTG-3′. The reactions of the p16 and GAPDH
gene amplification were performed under the following conditions:
95°C for 10 min; and 40 cycles of 95°C for 15 sec, 58°C for 40 sec
and 37°C for 1 min. The PCR profile of RAR-β consisted of 95°C for
10 min, 10 cycles of 95°C for 15 sec and 60°C for 30 sec, followed
by 40 cycles of 95°C for 10 sec and 58°C for 35 sec. The results
were analyzed using ABI Prism 7500 SDS software (Applied
Biosystems). The cycle threshold (CT) was determined and the
differences in the CT values for p16, RAR-β and GAPDH were
calculated. The expression of Genes with a CT of >35 cycles was
considered absent. p16 and RAR-β expression was normalized to the
simultaneously analyzed GAPDH. The comparative CT method was used
to determine the relative expression levels of p16 and RAR-β, and
the cycle number difference (ΔCT = CT p16/ RAR-β - CT GAPDH) was
calculated for each sample. The relative p16 and RAR-β expression
values are presented as 2(-ΔCT). Each sample was
measured in triplicate.
Western blot analysis
For the western blot analysis, the cells were
treated with DAC and ATRA for 48 h and then collected. Total
protein from the cell line was extracted using cell lysis buffer
for western blot anaylsis containing phenylmethanesulfonyl fluoride
(Beyotime) according to the manufacturer’s instructions. The
proteins were separated by 10% SDS polyacrylamide gel and
transferred to nitrocellulose membranes. Following blocking with 5%
skimmed milk, the membranes were incubated with an appropriate
dilution of the rabbit anti-human polyclonal primary antibodies,
anti-p16 (1:100), -RAR-β (1:100) and -GAPDH (1:1,000) (ABGENT, San
Diego, CA, USA), followed by incubation with the horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibody
(ABGENT), according to manufacturer’s instructions. The signals
were displayed using an automatic film processor (SX435-T; Taixing
Suxing Co., Ltd., Taixing, China). The signal intensity of the
proteins was normalized against GAPDH using Quantity One software
(Bio-Rad, Hercules, CA, USA).
Statistical analysis
All experiments were repeated three times with
similar results, and data are presented as the mean ± standard
deviation. Data were analyzed using the one-way analysis of
variance (ANOVA) and factorial design ANOVA. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using the statistical software
SPSS 17.0 (SPSS, Inc., Chicago, IL, USA).
Results
DAC and ATRA, alone or in combination,
have no effect on the growth inhibition, differentiation and
apoptosis of K562 cells
The inhibitory rates of the drug-treated cells were
all <20% (data not shown) and therefore, DAC and ATRA, alone or
in combination, had no effect on growth inhibition (P>0.05).
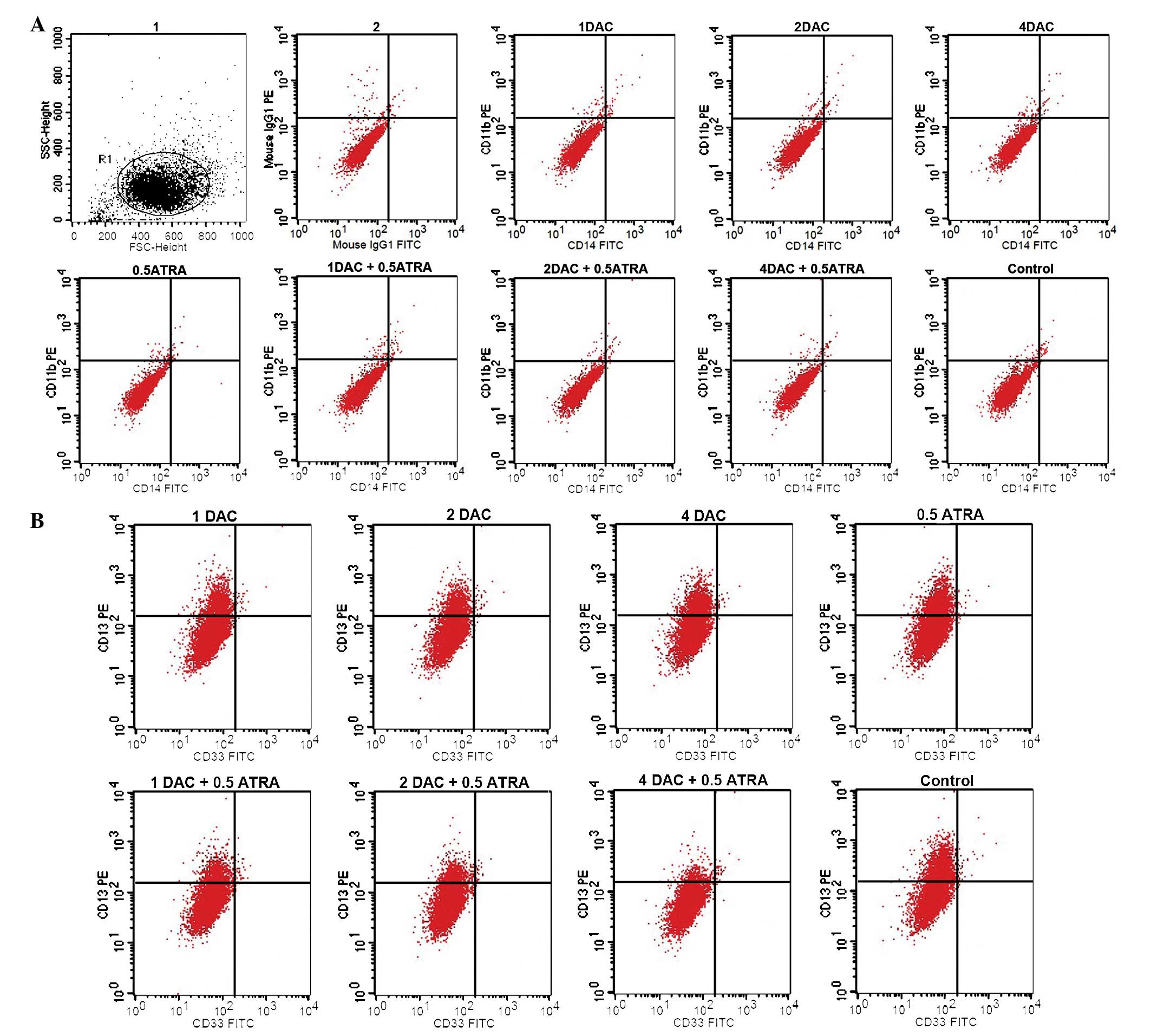
When the K562 cells were treated with the two drugs, no significant
induction of CD11b or CD14 expression (typical markers of
myelomonocytic differentiation of leukemia cells) was observed. The
expression level of CD13 was 17.28±0.79% in the group treated with
1 μmol/l DAC plus 0.5 μmol/l ATRA, 12.56±0.81% in the group treated
with 2 μmol/l DAC plus 0.5 μmol/l ATRA was and 5.45±0.76% in the
group treated with 4 μmol/l DAC plus 0.5 μmol/l ATRA. These results
indicated that the combination of DAC and ATRA was able to decrease
CD13 expression compared with the control group (32.25±1.34%;
P<0.05). The expression of CD33 was only faintly detected in the
experimental groups (Fig. 1). CD13
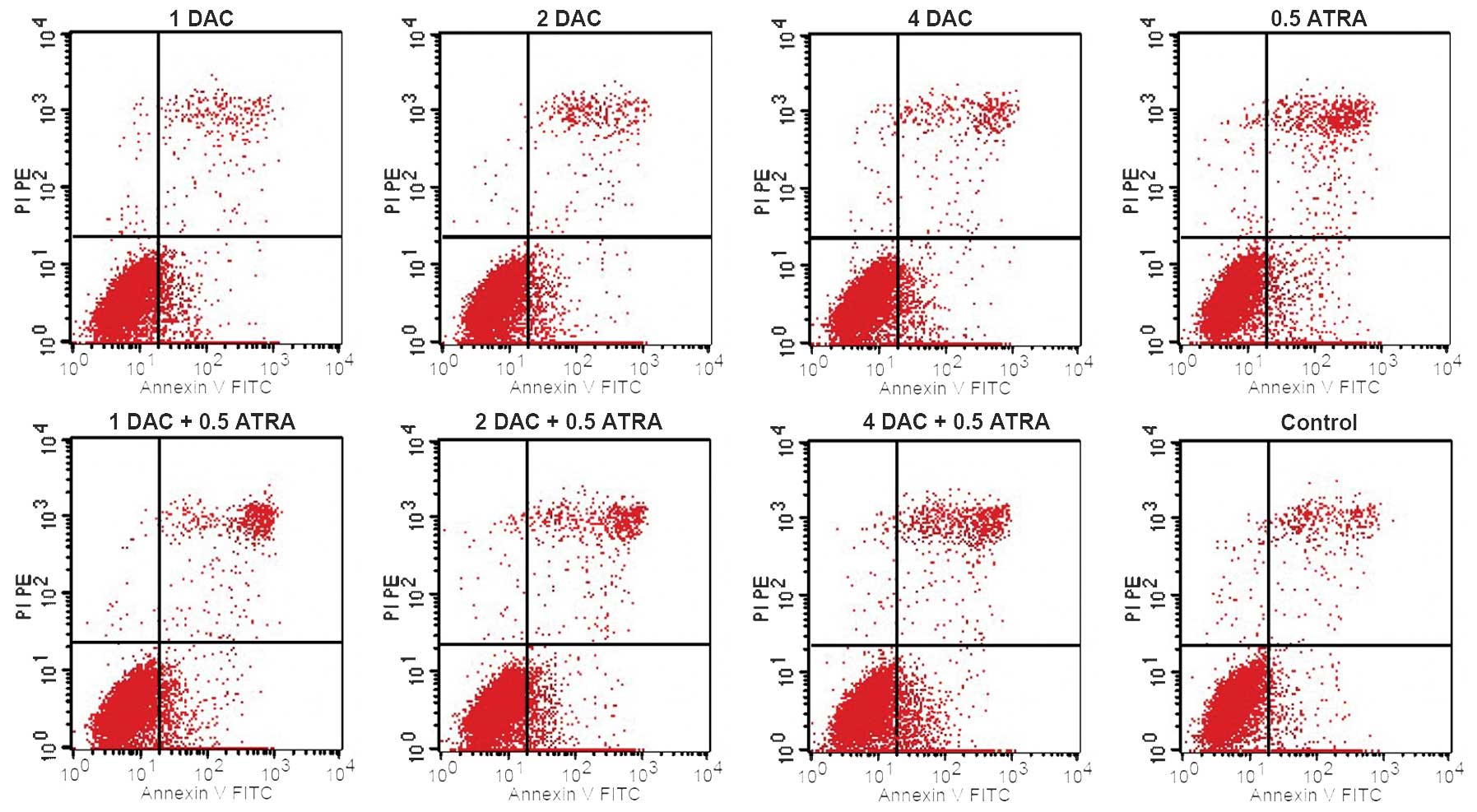
and CD33 are markers of immature myeloid cells. DAC and ATRA, alone
or in combination, had no significant effect on the early apoptosis
of the K562 cells compared with the control group (P>0.05;
Fig. 2).
Analysis of the methylation of p16 and
RAR-β genes in K562 cells
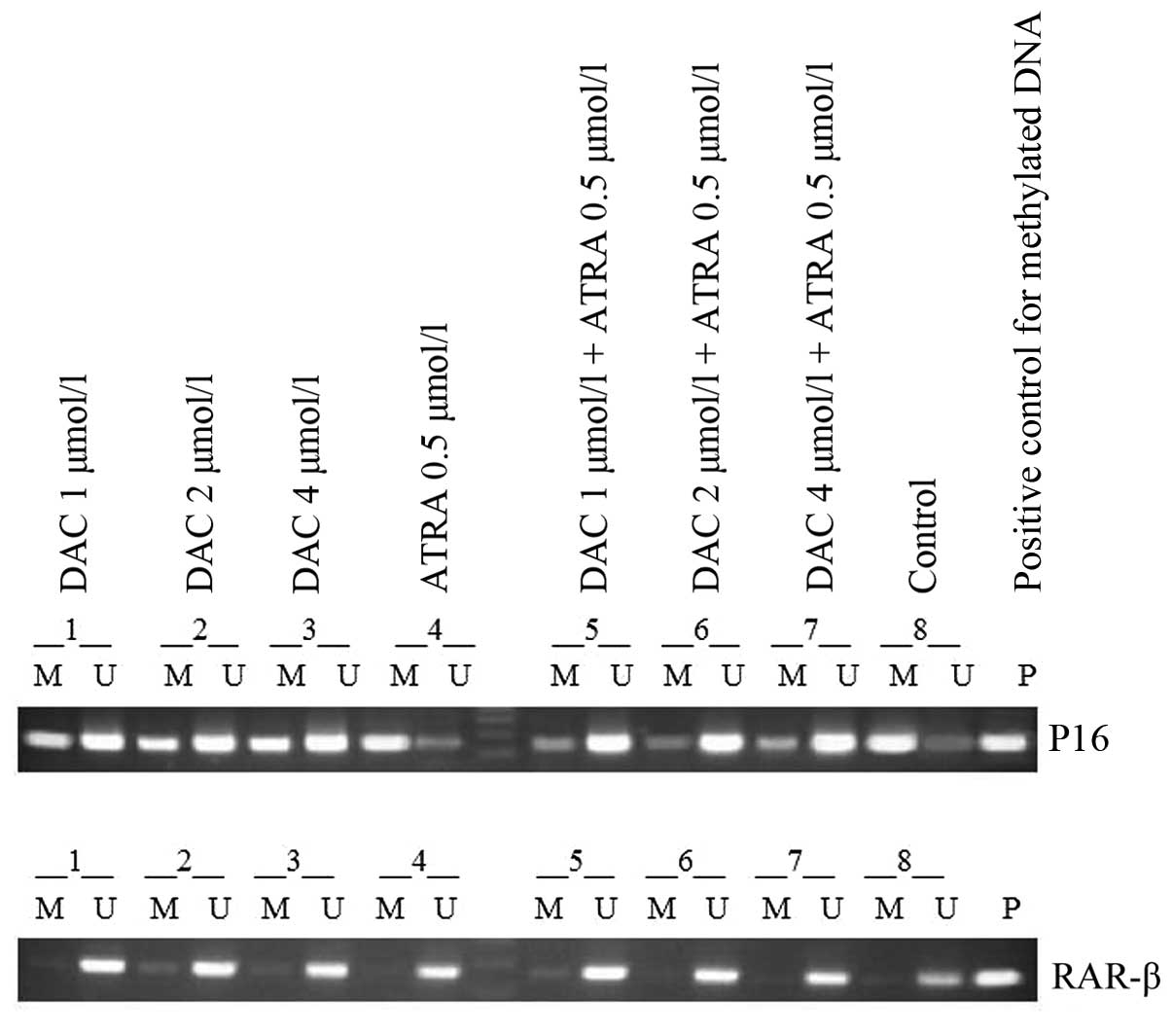
The MSP results showed clear signs of promoter
methylation in the K562 cells for p16, and partial demethylation
following treatment with DAC alone or in combination with ATRA, and
the combined effect was more evident than that of treatment with
DAC alone. The treatment with ATRA alone did not change the
methylation status of the DNA, whereas the K562 cell line exhibited
a band only when amplified with RAR-β primers for unmethylated and
not for methylated DNA (Fig.
3).
DAC in combination with ATRA activates
the mRNA expression of the RAR-β gene in K562 cells
The mRNA levels of the p16 and RAR-β genes were
detected by qPCR in K562 cells treated with drugs for 48 h. The
results indicated that p16 was silenced by aberrant methylation in
the K562 cells, and that DAC or ATRA, alone or in combination, were
unable to re-express p16 by the demethylation of DNA. However, the
RAR-β gene was unmethylated in the K562 cells, and the treatment of
the cells with a combination of DAC and ATRA resulted in the
upregulation of RAR-β. Following treatment of the cells with 1, 2
and 4 μmol/l DAC in combination with ATRA, the expression levels of
RAR-β were 0.000003±0.000002, 0.000048±0.000015 and
0.000405±0.000093, respectively (P<0.05).
Effect of DAC and ATRA on the protein
expression levels of p16 and RAR-β in K562 cells
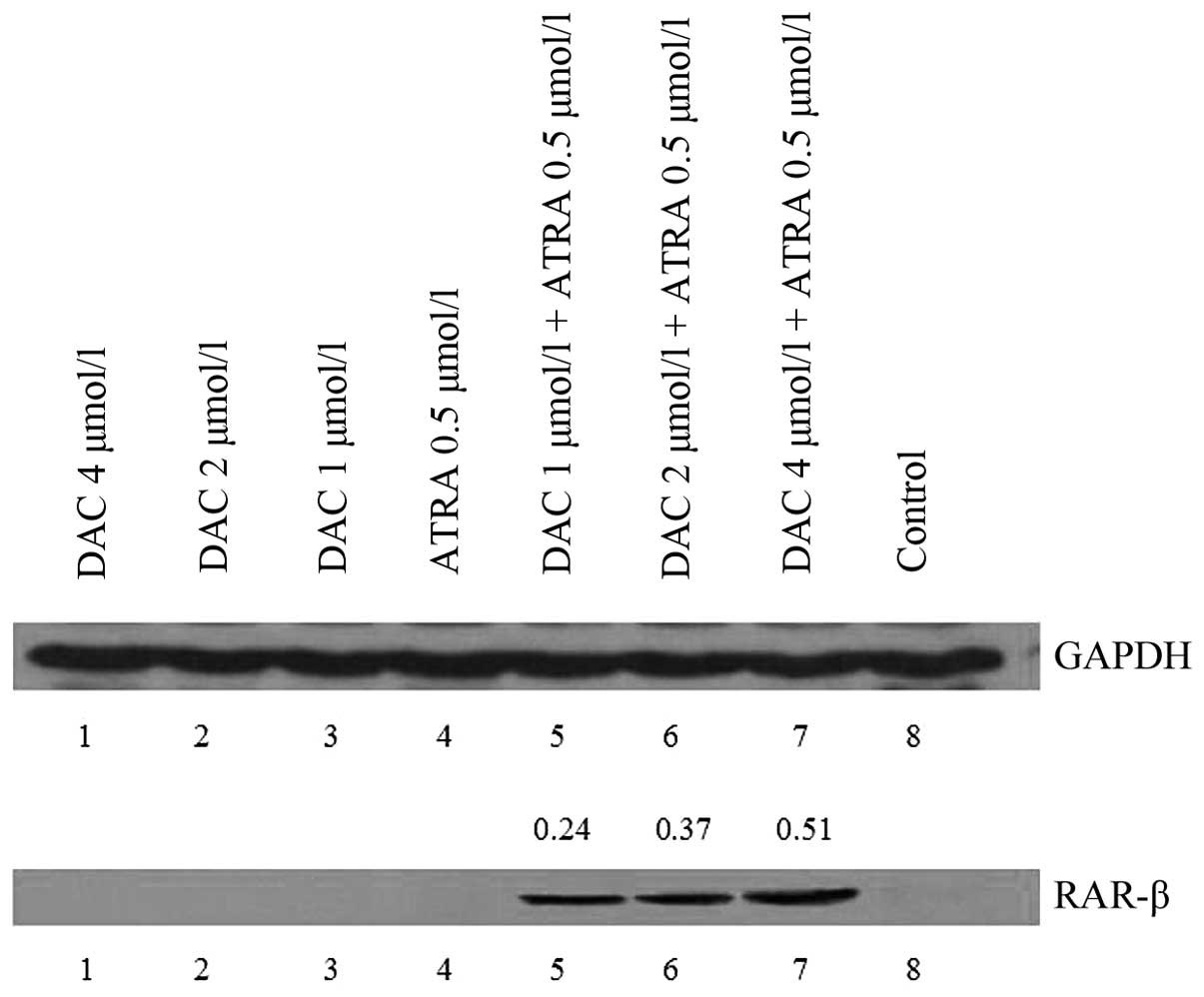
Western blot analysis was performed on the K562
cells treated with DAC and ATRA for 48 h. No visible bands of p16
and RAR-β protein were detected in the untreated samples, however,
RAR-β proteins were expressed in the drug-treated cells.
Furthermore, strong bands were observed at higher doses of DAC in
combination with ATRA (P<0.05; Fig.
4). The two drugs had no effect on p16 protein expression,
which correlates with the changes in the mRNA level observed by
qPCR.
Discussion
Previous studies have reported that DAC and ATRA
have the capacity to induce leukemic cell differentiation (10,12).
Furthermore, DAC inhibits esophageal squamous carcinoma cell and
thyroid carcinoma cell proliferation (13,14).
The induction of apoptosis is of particular interest as a potential
mechanism of action of the demethylating agents on the malignant
and pre-malignant hematopoietic cells in leukemia. Aoyama et
al (15) reported that the
differentiating and apoptotic effects of DAC were dependent on the
PU.1 expression level in PU.1-transgenic K562 cells. The current
study demonstrated that DAC and ATRA have no effect on the growth
inhibition, differentiation and apoptosis of K562 cells. However,
the combination of DAC and ATRA was able to decrease CD13
expression. These observations indicated that the differentiating
and apoptotic effects of DAC and ATRA in K562 cells may be
associated with the regulation of the expression of certain
genes.
The pathogenesis of myeloid malignancies involves
epigenetic changes, and efficacy has been shown by hypomethylating
agents in these diseases. The methylation of the 5′CpG island in
the p16 and RAR-β genes is associated with the transcriptional
silencing of the gene in a number of neoplasms, including solid
tumors and leukemias (16).
Uenogawa et al (17)
reported that azacitidine induces the demethylation of p16 in adult
T-cell leukemia/lymphoma. In the current study, the aim of the MSP
analysis of p16 and RAR-β in the K562 cells was to assess the
methylation status of the promoter in the genes. The results showed
clear signs of promoter methylation in the K562 cells for p16, and
partial demethylation was evident following the treatment of the
K562 cells with DAC alone or in combination with ATRA, but more
evidently for the combined effect. ATRA alone had no effect on
methylation and the RAR-β promoter region was not methylated in the
K562 cells. The K562 cells did not express p16 and RAR-β, however,
the expression of RAR-β was upregulated by treatment with DAC in
combination with ATRA for 48 h. A higher expression of RAR-β was
observed at higher doses of DAC in combination with ATRA, but the
two drugs had no effects on p16 expression. These results indicated
that DAC induces p16 promoter demethylation, however, the
demethylation does not affect the mRNA and protein expression of
p16. The RAR-β gene was unmethylated in the K562 cells, however,
DAC in combination with ATRA induced the expression of the tumor
suppressor gene, RAR-β, with unmethylated CpGs in the K562 cells.
Therefore, RAR-β may be one of the target genes of the two drugs in
acute erythroleukemia. We hypothesized that DAC upregulates RAR-β
gene expression in K562 cells, not by demethylation, but by any
other mechanism. However, the precise mechanism by which DAC in
combination with ATRA increases RAR-β expression in K562 cells has
not been well defined.
In conclusion, DAC induces p16 promoter
demethylation, and ATRA enhances this effect. The two drugs
synergistically activate RAR-β expression, which indicates that DAC
used in combination with ATRA has clinical potential in the
treatment of human erythroleukemia.
References
|
1
|
Issa J, Vertino PM, Boehm CD, et al:
Switch from monoallelic to biallelic human IGF2 promoter
methylation during aging and carcinogenesis. Proc Natl Acad Sci
USA. 93:11757–11762. 1996.
|
|
2
|
Melki JR and Clark SJ: DNA methylation
changes in leukaemia. Semin Cancer Biol. 12:347–357. 2002.
|
|
3
|
Takai D, Gonzales FA, Tsai YC, et al:
Large scale mapping of methylcytosines in CTCF-binding sites in the
human H19 promoter and aberrant hypomethylation in human bladder
cancer. Hum Mol Genet. 10:2619–2626. 2001.
|
|
4
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
et al; International Vidaza High-Risk MDS Survival Study Group.
Efficacy of azacitidine compared with that of conventional care
regimens in the treatment of higher-risk myelodysplastic syndromes:
a randomised, open-label, phase III study. Lancet Oncol.
10:223–232. 2009.
|
|
5
|
Steensma DP, Baer MR, Slack JL, et al:
Multicenter study of decitabine administered daily for 5 days every
4 weeks to adults with myelodysplastic syndromes: the alternative
dosing for outpatient treatment (ADOPT) trial. J Clin Oncol.
27:3842–3848. 2009.
|
|
6
|
Voso MT, Santini V, Finelli C, et al:
Valproic acid at therapeutic plasma levels may increase
5-azacytidine efficacy in higher risk myelodysplastic syndromes.
Clin Cancer Res. 15:5002–5007. 2009.
|
|
7
|
Merlo A, Herman JG, Mao L, et al: 5′ CpG
island methylation is associated with transcriptional silencing of
the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med.
1:686–692. 1995.
|
|
8
|
Herman JG, Jen J, Merlo A and Baylin SB:
Hypermeth- ylation-associated inactivation indicates a tumor
suppressor role for p15INK4B. Cancer Res. 56:722–727. 1996.
|
|
9
|
Côté S, Sinnett D and Momparler RL:
Demethylation by 5-aza-2′-deoxycytidine of specific
5-methylcytosine sites in the promoter region of the retinoic acid
receptor beta gene in human colon carcinoma cells. Anticancer
Drugs. 9:743–750. 1998.
|
|
10
|
Breitman TR, Selonick SE and Collins SJ:
Induction of differentiation of the human promyelocytic leukemia
cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA.
77:2936–2940. 1980.
|
|
11
|
Honma Y, Takenaga K, Kasukabe T and Hozumi
M: Induction of differentiation of cultured human promyelocytic
leukemia cells by retinoids. Biochem Biophys Res Commun.
95:507–512. 1980.
|
|
12
|
Momparler RL, Bouchard J and Samson J:
Induction of differentiation and inhibition of DNA methylation in
HL-60 myeloid leukemic cells by 5-AZA-2′-deoxycytidine. Leuk Res.
9:1361–1366. 1985.
|
|
13
|
Liu Z, Zhang L, Ding F, et al:
5-Aza-2′-deoxycytidine induces retinoic acid receptor-beta(2)
demethylation and growth inhibition in esophageal squamous
carcinoma cells. Cancer Lett. 230:271–283. 2005.
|
|
14
|
Miasaki FY, Vivaldi A, Ciampi R, et al:
Retinoic acid receptor beta2 re-expression and growth inhibition in
thyroid carcinoma cell lines after 5-aza-2′-deoxycytidine
treatment. J Endocrinol Invest. 31:724–730. 2008.
|
|
15
|
Aoyama S, Nakano H, Danbara M, et al: The
differentiating and apoptotic effects of 2-aza-5′-deoxycytidine are
dependent on the PU.1 expression level in PU1-transgenic K562
cells. Biochem Biophys Res Commun. 420:775–781. 2012.
|
|
16
|
Herman JG, Civin CI, Issa JP, et al:
Distinct patterns of inactivation of p15INK4B and p16INK4A
characterize the major types of hematological malignancies. Cancer
Res. 57:837–841. 1997.
|
|
17
|
Uenogawa K, Hatta Y, Arima N, et al:
Azacitidine induces demethylation of p16INK4a and inhibits growth
in adult T-cell leukemia/lymphoma. Int J Mol Med. 28:835–839.
2011.
|