Introduction
Multidrug-resistance (MDR) is the major complication
and a formidable obstacle in the therapy of acute leukemia (AL).
Although allogeneic hematopoietic stem cell transplantation (HSCT)
is a highly effective treatment for leukemia, its therapeutic
potential is counterbalanced by treatment-related toxicity and
graft-versus-host disease (GVHD). Therefore, the development of
therapy to reverse MDR is extremely important in the therapy of AL
(1,2).
Immunotherapy, which stimulates a patient’s own
immune system, is a promising method to overcome the drug
resistance to chemotherapy. Cytokine-induced killer (CIK) cells are
major histocompatibility complex (MHC)-unrestricted cytotoxic
lymphocytes generated with tumor necrosis factor-α (TNF-α),
interferon-γ (IFN-γ), interleukin (IL)-2 and IL-12. The activity of
CIK cells with regard to tumor cells is effective, moderate and
MHC-unrestricted. This activity is mainly associated with the high
proliferation potential of CIK cells (3,4)
In the present study, CIK cells were obtained from
the peripheral blood of healthy donors. The K562/ADR cells were
cultured with Adriamycin following incubation with CIK cells. The
study analyzed the effects of ADR following incubation with CIK on
reversing MDR in the K562/ADR cells and clarified its
mechanism.
Materials and methods
Cell culture
Human cell lines (K562/ADR and K562) were preserved
by tje International Medical Center of the First Central Hospital
of Tianjin (Tianjin, China) and cultured in RPMI-1640 complete
medium (Cambrex Bio Science, Verviers, Belgium) containing 10% heat
inactivated fetal calf serum (FCS), 100 U/ml penicillin and 100
mg/ml streptomycin. Prior to the study, the K562/ADR cells were
cultured in complete culture solution without Adriamycin.
Generation of CIK cells
Human peripheral blood mononuclear cells were
isolated from six healthy donors by Ficoll-Paque density
centrifugation (GE Healthcare, Fairfield, CT, USA) at 500 × g for
20 min and washed three times with phosphate-buffered saline (PBS).
The final cells were resuspended at a density of 3×106
cells/ml in RPMI-1640 complete medium containing 10% heat
inactivated FCS, 100 U/ml penicillin and 100 mg/ml streptomycin,
and were seeded at 37°C, 5% CO2. To generate CIK cells,
IFN-γ (1×106 U/l) was added on day 1 and rhIL-1
(1×105 U/l) and rhIL-2 (1×106 U/l) were added
on day 2. Fresh complete medium with rhIL-2 (1×106 U/l)
was added every 2–3 days, and the cells were harvested on day
14.
Cytokine secretion assays
Cytokine secretion by the CIKs was detected by
enzyme-linked immunosorbent assay (ELISA; R&D Systems,
Emeryville, CA, USA) where 3×105 cells were seeded into
a 6-well microplate and incubated overnight. The medium without FCS
was added, and the secretion of TNF-α, IFN-γ, IL-2 and IL-12 was
measured 72 h later following the manufacturer’s instructions.
Cytotoxicity assays
MTS cytotoxicity assays were used to determine the
viability and proliferation of the K562/ADR cells. The K562/ADR
cells were divided into five groups: K562 cells (Parent group),
K562/ADR cells (Group I), K562/ADR with CIK (Group II) and K562/ADR
with ADR in combination with CIK (Group III). The effect
(CIK)/target (K562/ADR) ratio (E/T ratio) was 10:1 and 20:1 in
groups II and III. The concentration of ADR was 0, 0.1, 0.5, 1, 5
and 10 μg/ml in the parent group, group I and group III. The cells
of all groups were seeded at a density of 5×104 cells/ml
in 96-well plates with RPMI-1640 complete medium (100 μl/well) at
37°C in a 5% CO2 humidified atmosphere. Next, different
E/T ratios (10:1 or 20:1) or ADR was added for 72 h. The cells were
incubated for 2 h at 37°C, 5% CO2 with MTS agent
(Promega Corporation, Madison, WI, USA) and the cytotoxicity of the
K562/ADR and K562 cells was measured at 570 nm.
Flow cytometry assays
The cells were cultured in a 6-well plate for 24 h,
then incubated with different ratios of CIK (10:1 or 20:1) for 72
h. The cells were then digested, resuspended, incubated with P-gp
antibodies for 30 min at 4°C and washed twice in PBS. The
fluorescence intensity of fluorescein isothiocyanate (FITC)-P-gp
(Abcam, Burlingame, CA, USA) was analyzed by flow cytometry
(FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA) at 488
nm.
The cells were cultured in a 6-well plate for 24 h,
then incubated with different ratios of CIK for 72 h. The cells
were then digested, resuspended, incubated with 10 μM Rh-123
(Sigma-Aldrich, San Francisco, CA, USA) for 60 min and washed twice
in PBS. The fluorescence intensity of Rh-123 was analyzed by flow
cytometry (FACSCalibur; BD Biosciences) at 488 nm.
The cells were cultured in a 6-well plate for 24 h,
then incubated with different ratios of CIK (10:1 or 20:1) for 72
h. A total of 10 μg/ml ADR was added and co-cultured for 24 h, then
the cells were digested, resuspended, incubated with Annexin V-FITC
and propidium iodide (PI) for 15 min at 37°C and washed twice in
PBS. The apoptosis rate was analyzed by flow cytometry
(FACSCalibur; BD Biosciences) at 488 nm.
GSH determination assays
The cells were cultured in a 6-well plate for 24 h,
then incubated with different ratios of CIK (10:1 or 20:1) for 72
h. Next, the cells were digested, resuspended and lysed.
Intracellular GSH was measured the by the Total GSH Assay kit
(Beyotime Institute of Biotechnology, Shanghai, China), according
to the manufacturer’s instructions, using a Spectra Max M5
microplate reader (Molecular Devices Corporation, Sunnyvale, CA,
USA).
Western blotting assays
The cells were cultured in a 6-well plate for 24 h,
then incubated with different ratios of CIK (10:1 or 20:1) for 72
h. The cells were then digested, resuspended and lysed. Next,
centrifugation at 10,000 × g was performed for 10 min at 4°C, and
the supernatant was extracted to obtain the total protein.
Electrophoresis was performed in 12% SDS polyacrylamide gel and the
protein was transferred to polyvinylidene fluoride membranes. The
membranes were blocked by 5% skimmed milk overnight at 4°C, then
monoclonal rabbit anti-human MDR1, MRP1, GST-π, Bcl-2, Survivin,
p-AKT and β-actin antibodies (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) were added and incubated at 4°C overnight. The
membranes were washed and incubated for 1 h with peroxidase-labeled
anti-rabbit immunoglobulin G. Finally, the membranes were exposed
to the Immobilon™ western chemiluminescent horseradish peroxidase
substrate for 1 min and visualized.
Real-time PCR analysis
The cells were cultured in a 6-well plate for 24 h,
then incubated with different ratios of CIK (10:1 or 20:1) for 72
h, and digested, resuspended and lysed. The total mRNA was
extracted and reverse transcribed. The transcription levels of
MDR1, MRP1 and GST-π, were detected by semiquantitative real-time
PCR using the icycler iQ detection system (Bio-Rad, Hercules, CA,
USA). The PCR conditions were as follows: Decontamination at 50°C
for 60 sec, then denaturation at 95°C for 40 sec, followed by 40
cycles at 95°C for 20 sec and hybridization at 95°C for 30 sec. The
oligonucleotide sequences were as follows: MDR1 forward, 5′-AAA
AAGATCAACTCGTACCACTC-3′, and reverse, 5′-GCACAAAATACACCAACAA-3′;
MRP1 forward, 5′-ACTTCCACATCTGCTTCGTCAGTG-3′ and reverse,
5′-ATTCAGCCACAGGAGGTAGAGAGC-3′; GST-π forward,
5′-TGGGCATCTGAAGCCTTTTG-3′ and reverse, 5′GATCTGGTCACCCAC
GATGAA-3′; Bcl-2 forward, 5′-ACGGGGTGAACTGGGGGAGGA-3′ and reverse,
5′-TGTTTGGGGCAGGCATGTTGACTT-3′; and Survivin forward,
5′-AGAACTGGCCCTTCTTGGAGG-3′ and reverse,
5′-CTTTTTATGTTCCTCTATGGGGTC-3′. GAPDH was used for normalization:
Forward, 5′-AACTTTGGCATTGTGGAAGG-3′ and reverse,
5′-ACACATTGGGGGTAGGAACA-3′.
Reporter gene assay
The cells were cultured in a 6-well plate for 24 h
and 0.1 μg pGL 4.32[luc2P/NF-kB-RE/Hypro] and pGL
4.32[luc2P/AP-1-RE/Hypro] plasmids were transfected by
Lipofectamine 2000 for 6 h. Next, the cells were incubated with
different ratios of CIK (10:1 or 20:1) for 72 h, and luciferase
activity was measured the by the Dual-Glo Luciferase assay system
(Promega Corporation), according to the manufacturer’s
instructions, using a Spectra Max M5 microplate reader.
Statistical analysis
All data are expressed as the mean ± standard
deviation. Statistical analysis was performed using SPSS 12.0
(SPSS, Inc., Chicago, IL, USA). The differences between repeated
experiments in the three groups were computed using the Student’s
t-test and F test. P<0.05 was considered to indicate a
statistically significant difference.
Results
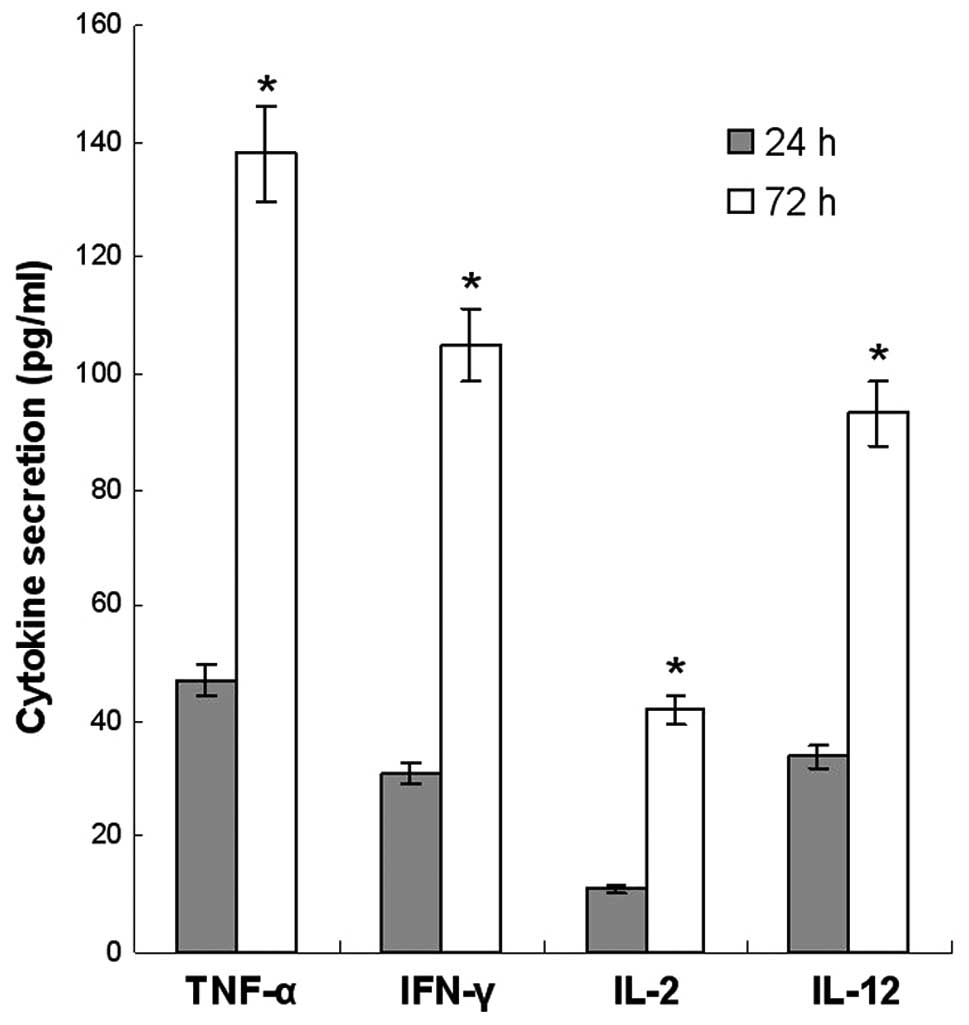
CIK cell cytokine secretion
Cytokine secretion by the CIK cells was detected by
ELISA assay. The CIK cells mainly produced IFN-γ, TNF-α, IL-2 and
IL-12 at the end of the culture period. There was an increased
secretion of these cytokines following 72 h compared with 24 h
(Fig. 1).
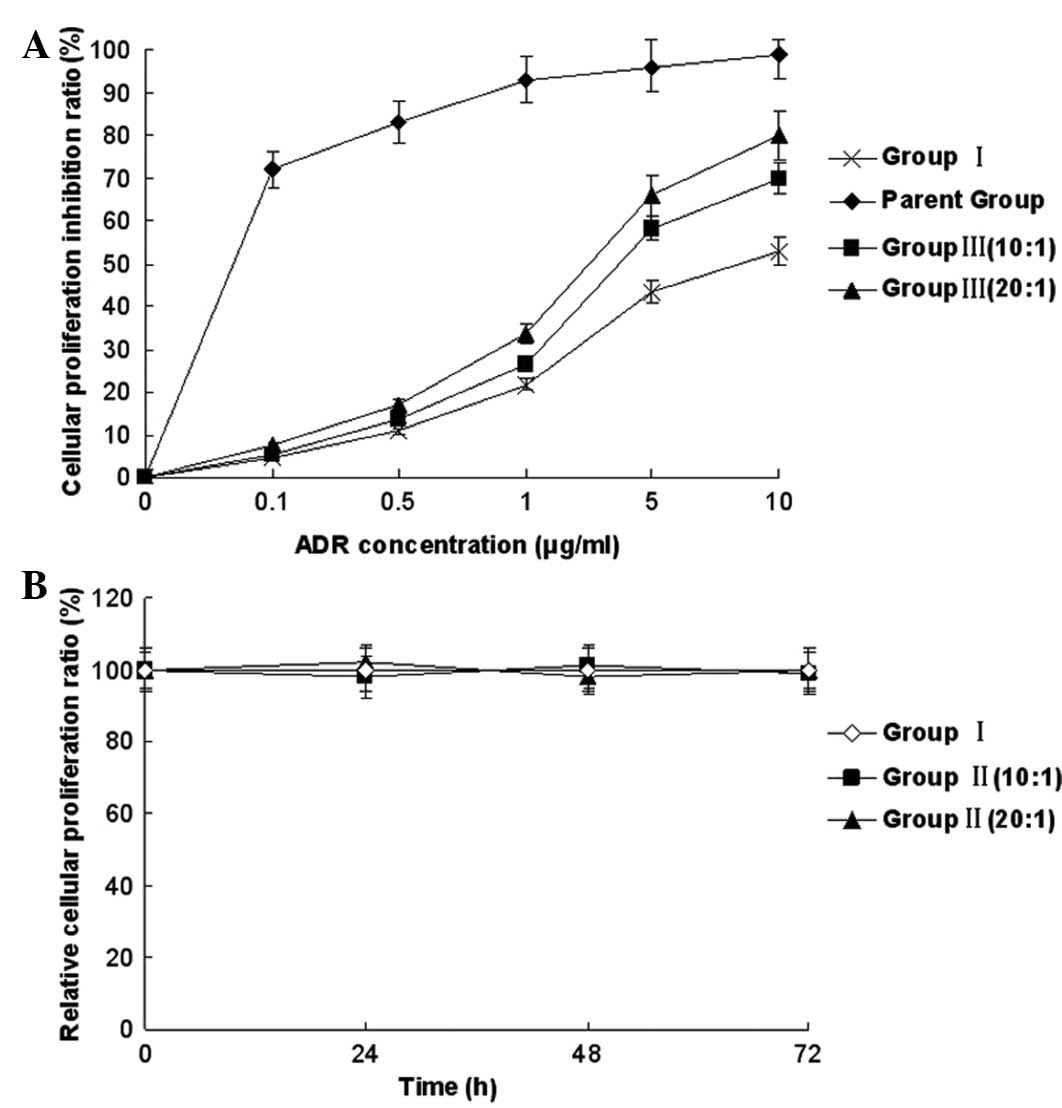
Effect and cytotoxic activity of CIK
cells on MDR reversal in K562/ADR and K562 cells
The cytotoxicity of the CIK cells was evaluated
against the K562/ADR and K5462 cell lines by MTS assay. With the
different ratio of effector and target cells, the cytotoxic
activity was determined. The cytotoxicity of group III at the
different ratios of E/T was higher than that of group I, indicating
the MDR reversal effect of CIK on the K562/ADR cells. Meanwhile,
the cytotoxicity of group II without ADR treatment was not
significantly changed compared with that of group I without ADR
treatment for 72 h, indicating that there was no significant
inhibitory effect of CIK on the K562/ADR cells (Fig. 2).
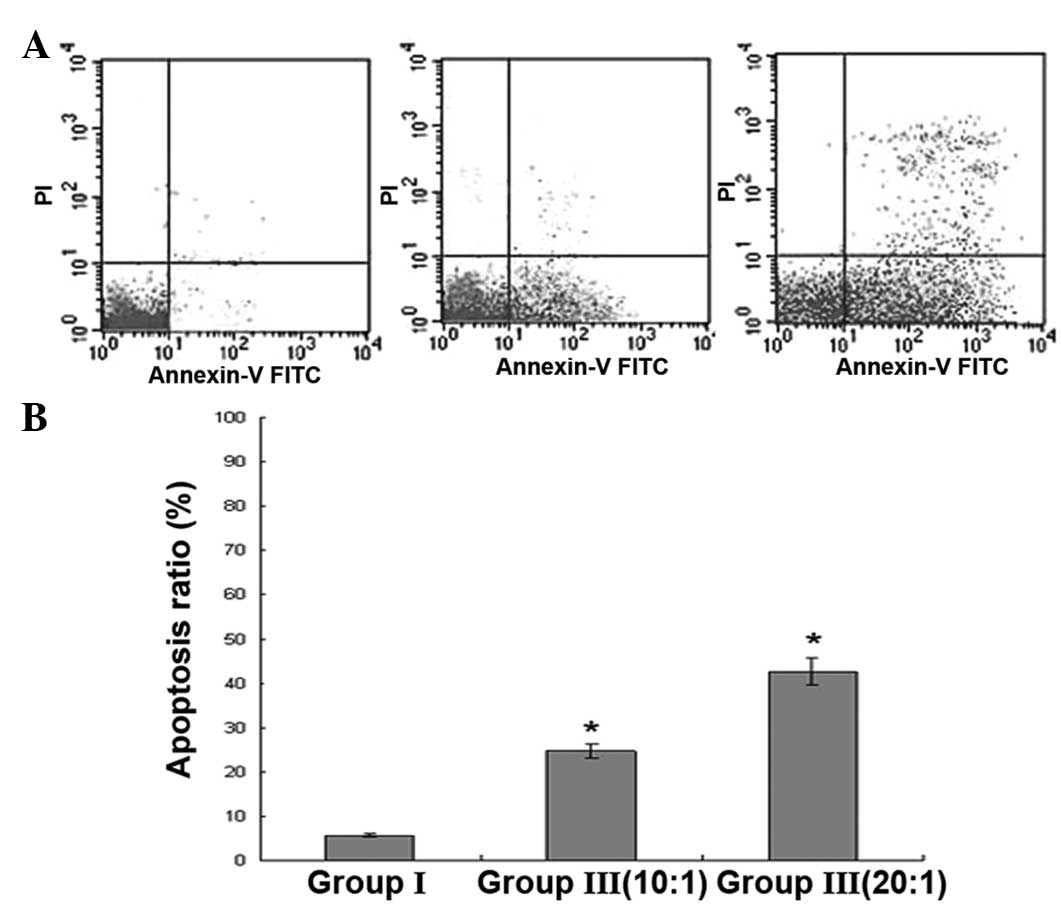
CIK-induced apoptosis of K562/ADR
cells
Apoptosis was detected by dual staining with Annexin
V-FITC and PI. The apoptosis rate of group III at the different
ratios of E/T was higher than that of group I. This revealed that
the increased apoptosis observed in group III could be induced by
the effect of CIK (Fig. 3).
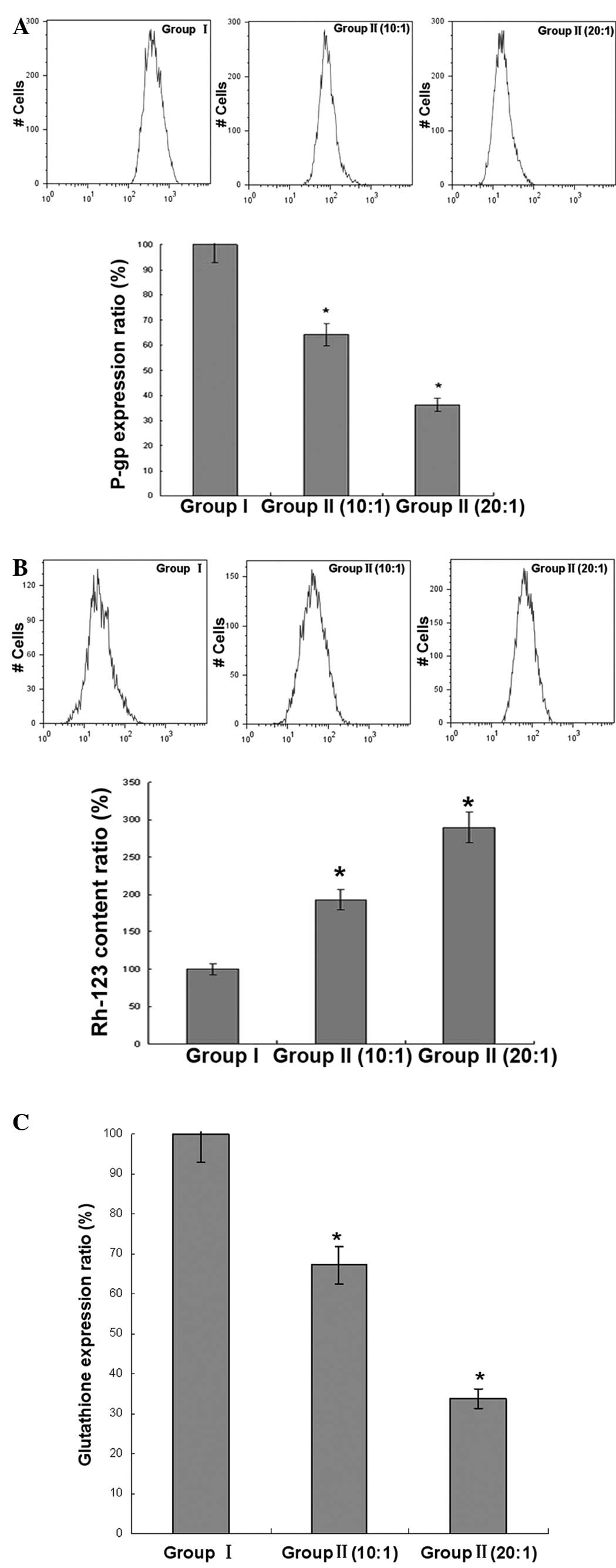
Intracellular content of Rh-123 and
expression of P-gp and GSH in K562/ADR cells
The intracellular Rh-123 content and expression of
P-gp was analyzed by flow cytometry. With the different ratios of
E/T, the intracellular Rh-123 content of group II was increased
compared with group I, and the expression of P-gp and GSH in group
II was lower than that of group I, indicating the increased effect
of the intracellular Rh-123 content of CIK and the inhibitory
effect of the expression of P-gp and GSH of CIK on the K562/ADR
cells (Fig. 4).
Expression of MDR-related gene in
K562/ADR cells
The expression of MDR1, MRP1, GST-π, Bcl-2 and
Survivin was analyzed by western blot and real-time PCR assays.
With the different ratios of E/T, the protein and mRNA expression
of these gene in group II was lower than that of group I,
indicating the inhibitory effect of the MDR-related gene expression
of CIK on the K562/ADR cells (Fig.
5).
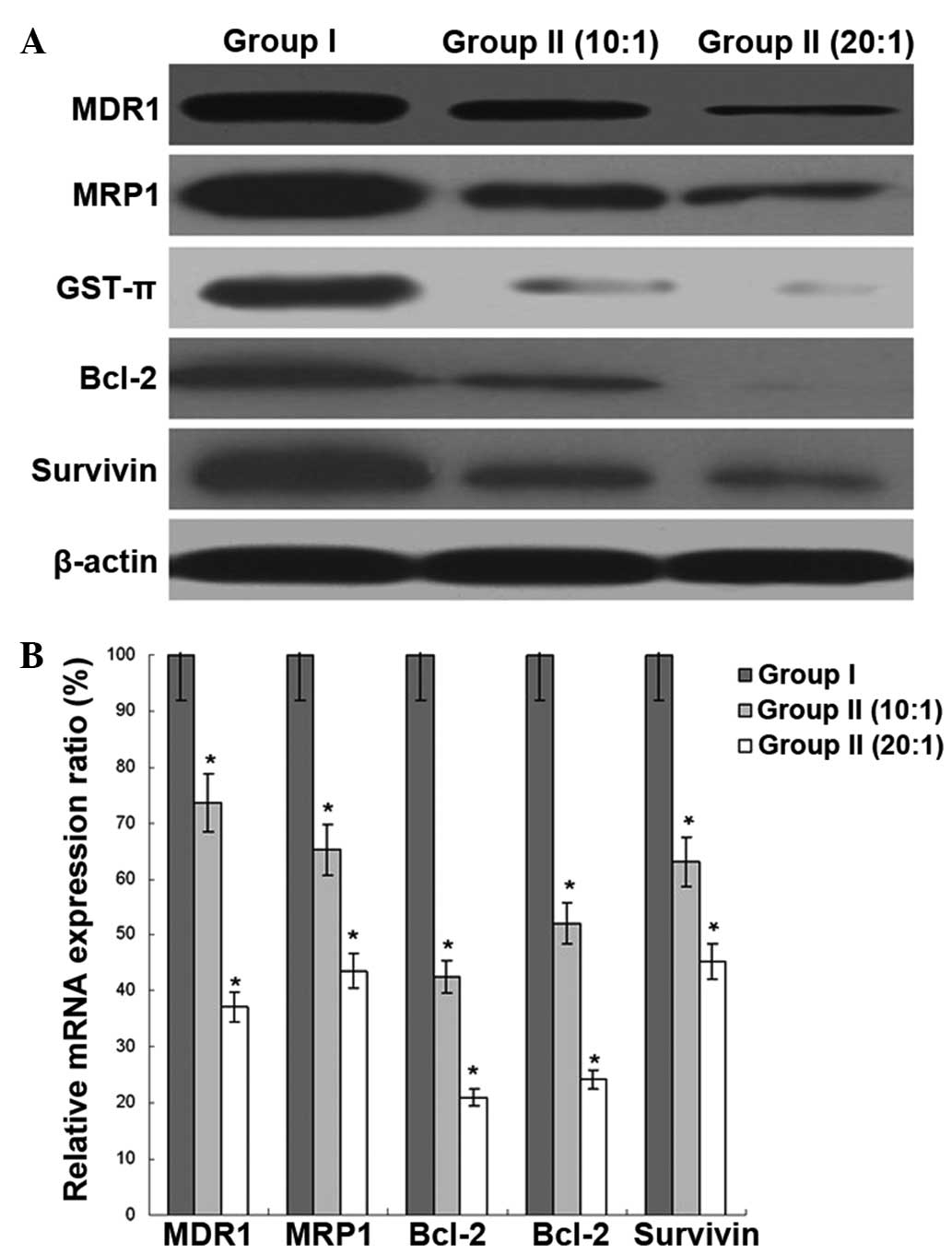 | Figure 5Effect of cytokine-induced killer
(CIK) cells on multidrug-resistance (MDR)-related gene expression
in K562/ADR cells. (A) The protein expression of MDR gene 1 (MDR1),
MDR-associated protein 1 (MRP1), GSH S-transferase-π (GST-π),
B-cell lymphoma 2 (Bcl-2) and Survivin was measured by western blot
assays in K562/ADR cells. β-actin was an internal reference. (B)
The mRNA expression of MDR1, MRP1, GST-π, Bcl-2 and Survivin was
measured by real-time PCR assays in K562/ADR cells. GAPDH was an
internal reference. The data was presented as the mean ± SD, n=5,
bars indicate SD, *P<0.05 vs. Group I. ADR,
Adriamycin. |
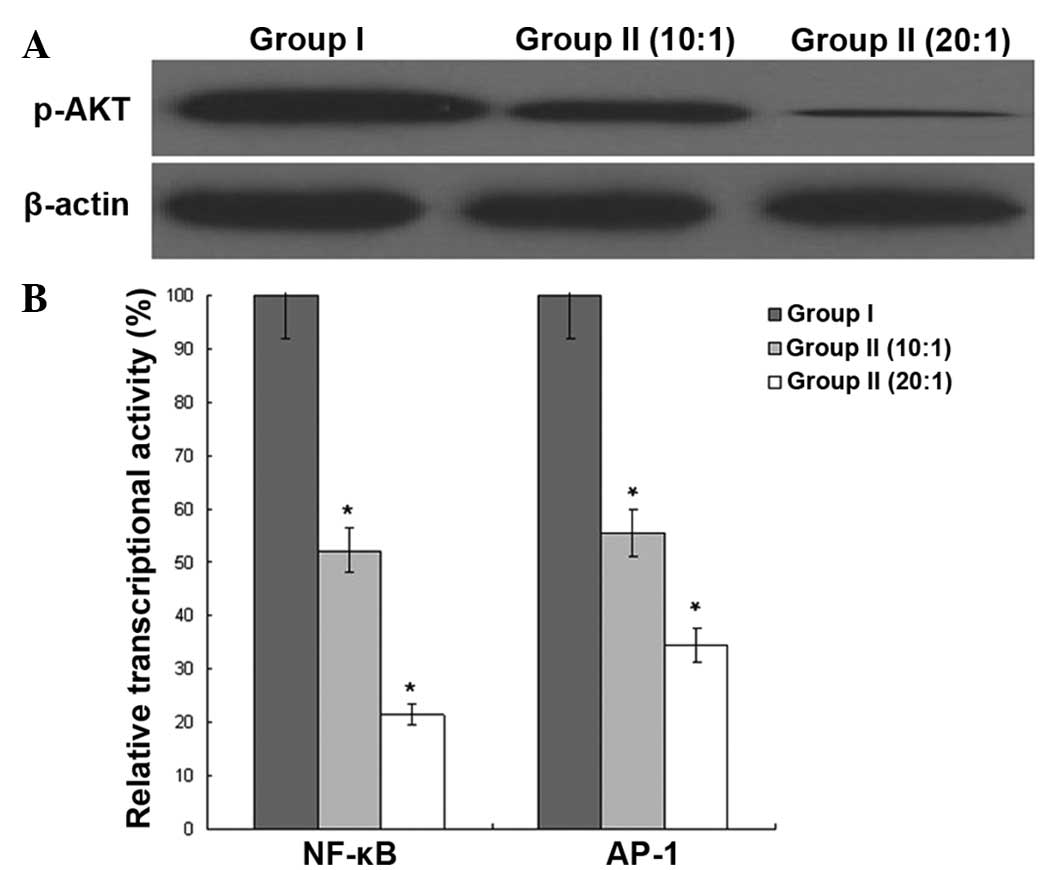
Regulation of signal transduction
molecules in K562/ADR cells
The phosphorylation of AKT was analyzed by western
blot assay, and the transcriptional activity of nuclear factor
(NF)-κB and activator protein 1 (AP-1) was analyzed by reporter
gene assay. With the different ratios of E/T, the phosphorylation
of AKT and the transcriptional activity of NF-κB and AP-1 of group
II were lower than that of group I, indicating the regulative
effect of the signal transduction molecules of CIK on the K562/ADR
cells (Fig. 6).
Discussion
Although allogeneic HSCT is a highly effective
treatment for leukemia, its therapeutic potential is
counterbalanced by treatment-related toxicity and GVHD (5). MDR is the major complication and a
formidable obstacle in the therapy of AL (6). Certain previous studies that used
cyclosporine and verapamil to reverse the MDR of AL were not
practical in the clinic due to the side-effects. Therefore, the
development of therapy for the absolute depletion of residual
leukemic cells is extremely important. In the present study, CIK
was acquired from the peripheral blood of healthy donors, and the
K562/ADR cells were cultured with Adriamycin following incubation
with CIK. It has been reported that MDR1, MRP1, GST-π, Bcl-2 and
Survivin are significant genes with respect to MDR in tumors
(7–9). The present study detected that the
cytotoxicity, intracellular Rh-123 content and apoptosis rate of
the K562/ADR cells was increased, while the expression of P-gp,
MDR1, MRP1, GST-π, Bcl-2 and Survivin was decreased by CIK
co-cultured with Adriamycin.
The CIK cells have anticancer activity in
vitro and in vivo by the production of effector
cytokines, such as IFN-γ, TNF-α, IL-2 and IL-12, which are involved
in immunoregulation (10). In the
present study, the pattern of cytokines in CIK cells was
characterized, including phenotype, cytokine secretion, cytotoxic
effects to K562/ADR cells. It was found that a lower E/T ratio
(10:1 and 20:1) could induce apoptosis and decrease the expression
of MDR-related genes in K562/ADR cells, although there was no
significant cytotoxicity with this ratio.
The phosphorylation of AKT could lead to the
transcriptional activity of NF-κB and AP-1 (11–13).
The present study found that CIK treatment led to downregulated AKT
phosphorylation, which meant that the PI3K/AKT pathway was
modulated by CIK. Meanwhile, further analysis confirmed that the
transcriptional activity of NF-κB and AP-1 was inhibited by CIK
treatment.
In summary, the present study provides evidence that
CIK has immunoregulatory and other functions in K562/ADR cells,
inducing the downregulation of the expression of MDR-related genes
and inducing apoptosis of the target cells. Overall, these
properties of CIK cells may be beneficial in the treatment of
leukemia, particularly for residual AL cells. The study conclusions
may provide useful tools for reversing the MDR of AL and be
practical in clinical application.
References
|
1
|
Ahn HK, Jang JH, Kim K, Kim HJ, Kim SH,
Jung CW and Kim DH: Monosomal karyotype in acute myeloid leukemia
predicts adverse treatment outcome and associates with high
functional multidrug resistance activity. Am J Hematol. 87:37–41.
2012.
|
|
2
|
Chauhan PS, Bhushan B, Singh LC, Mishra
AK, Saluja S, Mittal V, Gupta DK and Kapur S: Expression of genes
related to multiple drug resistance and apoptosis in acute
leukemia: response to induction chemotherapy. Exp Mol Pathol.
92:44–49. 2012.
|
|
3
|
Liu P, Chen L and Huang X: The antitumor
effects of CIK cells combined with docetaxel against drug-resistant
lung adenocarcinoma cell line SPC-A1/DTX in vitro and in vivo.
Cancer Biother Radiopharm. 24:91–98. 2009.
|
|
4
|
Deng Q, Bai X, Xiao X, Jiang Y and Li YM:
Reversion of multidrug resistance by CIK in K562/ADR cells and its
mechanism exploration. Zhonghua Xue Ye Xue Za Zhi. 32:52–56.
2011.(In Chinese).
|
|
5
|
Schuurhuis GJ, Zweegman S and Ossenkoppele
GJ: Highly effective mobilization of CD34 positive cells as a poor
prognostic factor in acute myeloid leukemia. Possible causes and
consequences. Leuk Res. 37:727–728. 2013.
|
|
6
|
van den Heuvel-Eibrink MM, Wiemer EA, de
Boevere MJ, Slater RM, Smit EM, van Noesel MM, van der Holt B,
Schoester M, Pieters R and Sonneveld P: MDR1 expression in
poor-risk acute myeloid leukemia with partial or complete monosomy
7. Leukemia. 15:398–405. 2001.
|
|
7
|
Li Y, Yan PW, Huang XE and Li CG: MDR1
gene C3435T polymorphism is associated with clinical outcomes in
gastric cancer patients treated with postoperative adjuvant
chemotherapy. Asian Pac J Cancer Prev. 12:2405–2409. 2011.
|
|
8
|
Matsunaga S, Asano T, Tsutsuda-Asano A and
Fukunaga Y: Indomethacin overcomes doxorubicin resistance with
inhibiting multi-drug resistance protein 1 (MRP1). Cancer Chemother
Pharmacol. 58:348–353. 2006.
|
|
9
|
Klaus A, Zorman S, Berthier A, Polge C,
Ramirez S, Michelland S, Sève M, Vertommen D, Rider M, Lentze N,
Auerbach D and Schlattner U: Glutathione S-transferases interact
with AMP-activated protein kinase: evidence for S-glutathionylation
and activation in vitro. PLoS One. 8:e624972013.
|
|
10
|
Tettamanti S, Marin V, Pizzitola I,
Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F,
Galimberti S, Lopez AF, Biondi A, Bonnet D and Biagi E: Targeting
of acute myeloid leukaemia by cytokine-induced killer cells
redirected with a novel CD123-specific chimeric antigen receptor.
Br J Haematol. 161:389–401. 2013.
|
|
11
|
Burris HA III: Overcoming acquired
resistance to anticancer therapy: focus on the PI3K/AKT/mTOR
pathway. Cancer Chemother Pharmacol. 71:829–842. 2013.
|
|
12
|
Gilmore TD: Introduction to NF-kappaB:
players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
|
|
13
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009.
|