Introduction
Mesenchymal chondrosarcoma (MCS), initially
described by Lichtenstein and Bernstein in 1959 (1), is one of the most unusual
chondrosarcomas, representing only 2–10% (2–5) of
these tumour types worldwide. MCS arises across a broad age
spectrum, generally between 20 and 40 years, but has also been
diagnosed in the paediatric population globally. This
chondrosarcoma type has been characterized as a high-grade tumour
with a propensity to metastasise to the lung, lymph nodes and bone
(6,7). MCS most frequently originates in the
bone, but is located in soft tissues in ~25% of cases, and is
occasionally detected adjacent to meninges and within the spinal
canal (8). Until recently, MCS has
lacked a specific diagnostic immunohistochemical profile or
consistent genetic alterations that facilitate its differentiation
from other bone tumours, and the diagnosis is generally based on
histological features, which vary considerably. The current
treatment of choice for MCS is surgery. To date, the efficacies of
adjuvant chemo- and radiotherapy remain poorly defined (9), but appear to improve clinical
outcomes. However, prognosis is extremely variable, as reflected in
the published 10-year overall survival rates, ranging from 21%
(10) to 67% (9). Improved understanding of the cell
biology of MCS would therefore present a major advantage in
accelerating the development of targeted drugs with enhanced
effectiveness for tumour treatment. Limited information is
currently available regarding the biology of MCS, with recorded
cases of intraspinal MCS being extremely rare.
Tumour-specific, balanced chromosomal translocations
have been identified in several histologically defined soft tissue
sarcomas over the last 20 years (11,12).
The first of these translocations was discovered in Ewing’s sarcoma
(13,14), and subsequent reports have
frequently demonstrated the specificity of these fusion genes
(15,16).
No consistent molecular markers have been
established for MCS until recently, although chromosomal reciprocal
translocations have been reported, such as (11;22)(q24;q12)
(17) and genetic findings of
trisomy 8 (17,18). In early 2012, a novel fusion gene,
HEY1-NCOA2, was identified in MCS (19). In the current study, we further
confirmed the presence of the recently identified HEY1-NCOA2 fusion
in a paediatric case of primary intradural MCS, supporting its
utility as a novel diagnostic marker for the disease. The study was
approved by the Regional Ethics Committee (Ethical Review Boards;
Gothenburg, Sweden) of the CWS Soft Tissue Tumour Registry and
written informed consent was obtained from the patient’s
parents.
Case report
Case report
A 10-year-old female presented to the Södra Älvsborg
Hospital (Borås, Sweden) with 9 months of back pain. General
physical examination revealed normal results. The patient was
referred to the Queen Silvia Children’s Hospital (Gothenburg,
Sweden) and the data from neurological tests, including mental
status, cranial nerve examination, cerebellar testing, and motor
and sensory tests of the lower extremities, were additionally
normal. No clinical evidence of a tumour was identified and the
patient had no family history of cancer or genetic disorders.
Magnetic resonance imaging (MRI) disclosed a 1.5-cm solid
intradural lesion at the level of Th4. Further laboratory tests
showed no abnormalities. The patient underwent surgery, with
macroscopically complete removal of a well-defined tumour attached
to the arachnoid roots, but not the dura mater or medulla spinalis.
Following recovery from surgery, the patient was subjected to
radiotherapy (proton radiation), specifically, 50.4 Gy in 1.8-Gy
fractions over a period of 6 weeks. The patient is currently free
of symptoms at two years following the completion of therapy. No
radiological findings of relapse have been detected following MRI
every four months.
Histological and immunohistochemical
analysis
All laboratory work, including morphological,
molecular pathological and immunohistochemical analysis, was
performed at the time of diagnosis. Resected tumour tissue was
fixed in 10% formalin, cut into small pieces and embedded in
paraffin. Tissue blocks were cut into 4-μm thick slices and stained
with haematoxylin and eosin.
For immunohistochemical analysis, the following
monoclonal primary antibodies were used: monoclonal rabbit
anti-human CD99 (Epitomics, Burlingame, CA, USA) diluted 1:1000,
monoclonal mouse anti-human Ki-67 [Flex Ready-to-Use (RTU) IR626;
Dako, Carpinteria, CA, USA] and monoclonal rabbit anti-human S-100
(Flex RTU IR504; Dako). Positive controls (S-100 for the wall of
appendix vermiformis; and CD99 and Ki-67 for the tonsils) were used
for all staining protocols, while the negative controls were
without primary antibodies. Slides were automatically stained using
a Dako Autostainer (LV-1 Autostainer; Dako). For CD99 staining,
slides were rehydrated with xylene, followed by a series of alcohol
dilutions. Antigen retrieval was performed using Tris-EDTA (pH 9.0)
in combination with heat induction in a microwave oven (8 min at
750 W, followed by 15 min at 350 W). Regarding Ki-67 and S-100
staining, the following procedure was used: PT Link (Pre-Treatment
Module for Tissue Specimens; Dako) for Ki-67 Target Retrieval
Solution, Low pH (K8005; Dako) and for S-100 Target Retrieval
Solution, High pH (K8004; Dako). Slides were rinsed twice in buffer
(EnVision™ FLEX Buffer; 8007; Dako), with subsequent blocking of
endogenous peroxidase by treatment with Peroxidase-Blocking
Solution (S2023; Dako) for 7 min, followed by incubation with
primary antibodies at room temperature for 25 min. Next, slides
were re-washed in buffer, and treated with the secondary antibody,
EnVision Flex, High pH (Link) (K800; goat anti-mouse/human
polyclonal; Dako) for Ki-67 and S-100 and Real EnVision (K5007;
horseradish peroxidase; goat anti-mouse/rabbit polyclonal; DAKO).
After 25 min of incubation and two further rinses with buffer for
CD99, slides were covered with the visualization agent, DAB (K3468;
Dako) for 10 min, rinsed, counterstained in EnVision Flex
Haematoxylin and mounted. All cells that showed immunoreactivity
for S-100 in the cartilage and CD99 in the spindle cell component
were counted at high power fields (magnification, ×400) using a
Nikon Eclipse E800 microscope (Nikon Corporation, Tokyo,
Japan).
RNA extraction, reverse
transcription-polymerase chain reaction (RT-PCR) and
sequencing
Paraffin-embedded tumour tissue was histologically
analysed, and a representative block was selected for the presence
of viable tumour cells selected. Thirty 5-μm sections were used for
total RNA extraction, using the RNeasy FFPE kit (Qiagen, Hilden,
Germany). The concentration of extracted total RNA was determined
with a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham,
MA, USA).
An aliquot of 300 ng total RNA was
reverse-transcribed and amplified using the Qiagen OneStep RT-PCR
kit (Qiagen). RT-PCR was conducted using the following primer set:
HEY1 forward (exon 4), 5′-ACCGTGGATCACCTGAAAAT-3′ and NCOA2 reverse
(exon 13), 5′-TGCAATGTGATGTCAAGTGG-3′, at an annealing temperature
of 61°C and for 40 cycles. The primers amplified a 119-bp product
representing a fragment of HEY1 exon 4 fused in-frame to NCOA2 exon
13 was amplified. As a positive control for the RNA integrity,
RT-PCR for the housekeeping gene, β-actin, was performed using the
following primer set: β-actin forward, 5′-ATCACCATTGGCAATGAGCG-3′
and reverse, 5′-TTGAAGGTAGTTTCGTGGAT-3′, at an annealing
temperature of 61°C and for 40 cycles. These primers amplified a
100-bp fragment of the housekeeping gene β-actin. An aliquot of the
amplified RT-PCR products was visualised by electrophoresis on a 2%
agarose gel stained with ethidium bromide.
The amplified RT-PCR product was purified using
Illustra Microspin S-300 HR columns (GE Healthcare, Ltd., Chalfont
St. Giles, United Kingdom). To confirm the presence of the fusion
transcript, the purified product was sequenced using the BigDye
Terminator v1.1 Cycle Sequencing kit and resolved on a 3130xl
Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Subsequently, sequences were manually aligned with the original
sequences of HEY1 (NM_012258.3) and NCOA2 (NM_006540.2) for
comparison.
Histological and immunohistochemical
results
Macroscopically, the tumour was chondromatous with
bone fragments displaying a grey-white and pink colour with
soft-to-firm consistency. No necrosis or haemorrhage was observed.
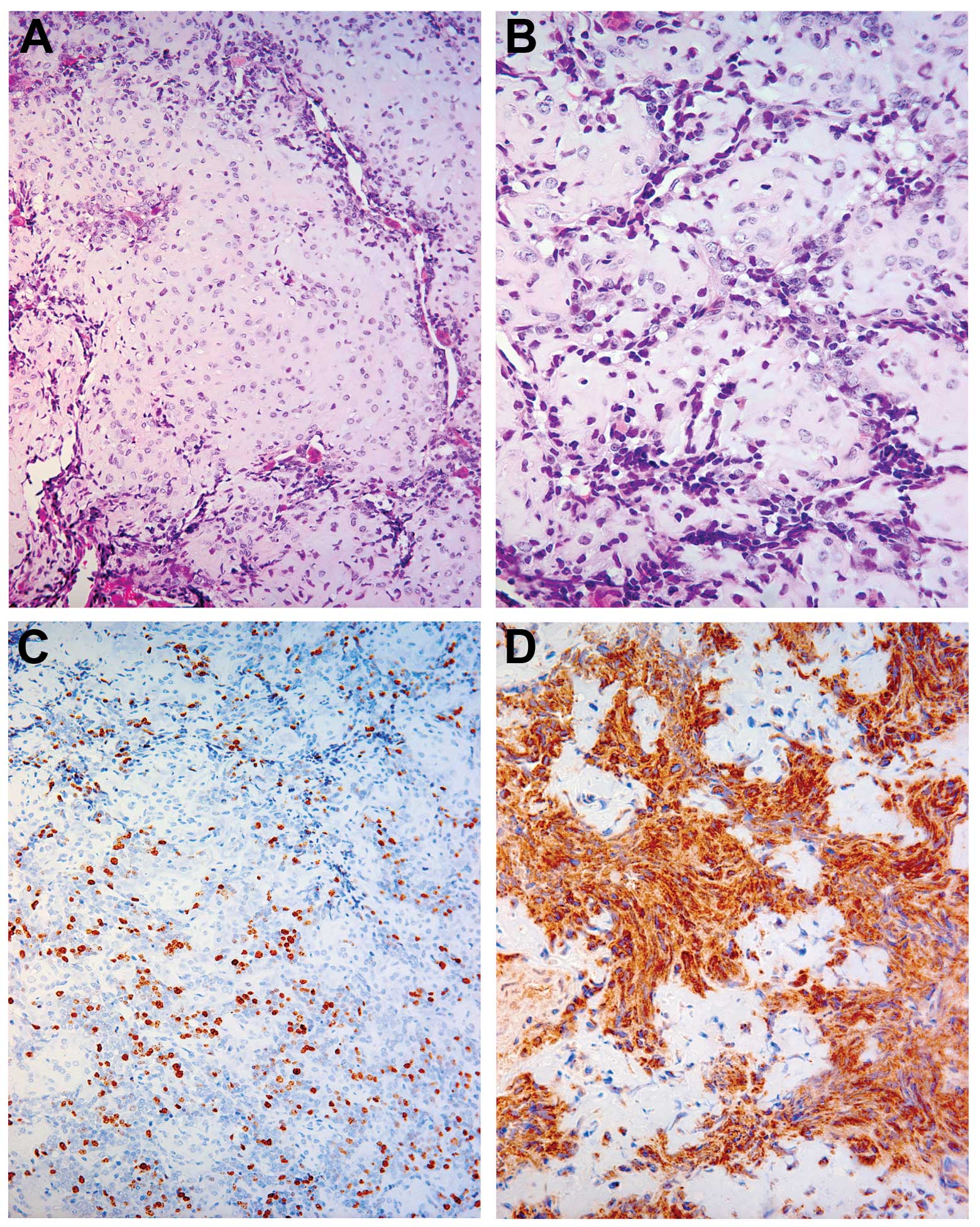
Microscopically, the tumour showed a biphenotypical appearance,
with both hypercellular and hypocellular areas. In terms of
cellular components, the tumour showed undifferentiated small
round-to-ovoid shaped cells with brisk mitotic activity and
hyperchromasia. Hypocellular islands with chondroid tissue and
well-differentiated hyaline cartilage were observed (Fig. 1A and B). Immunohistochemical
analysis disclosed that all undifferentiated cells stained positive
for CD99 (Fig. 1D), with a high
degree of positivity for Ki-67 (25%) (Fig. 1C) and for S-100 (100%) in the
cartilage component (data not shown).
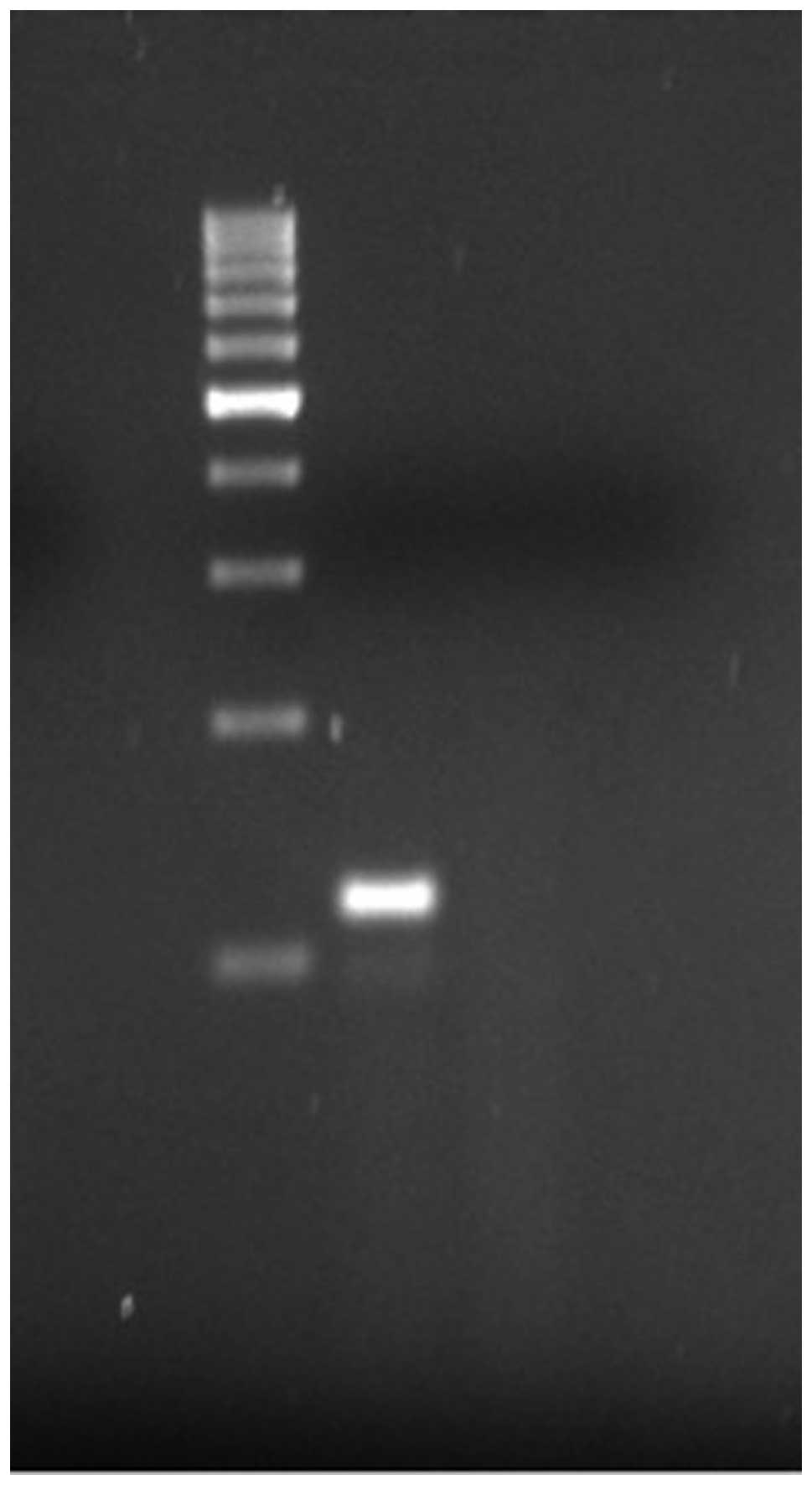
RT-PCR results
RT-PCR for the HEY1-NCOA2 fusion gene was attempted,
as paraffin-embedded tumour material was available. A strong 119-bp
band was obtained (Fig. 2) upon
amplification using a forward primer derived from exon 4 of HEY1
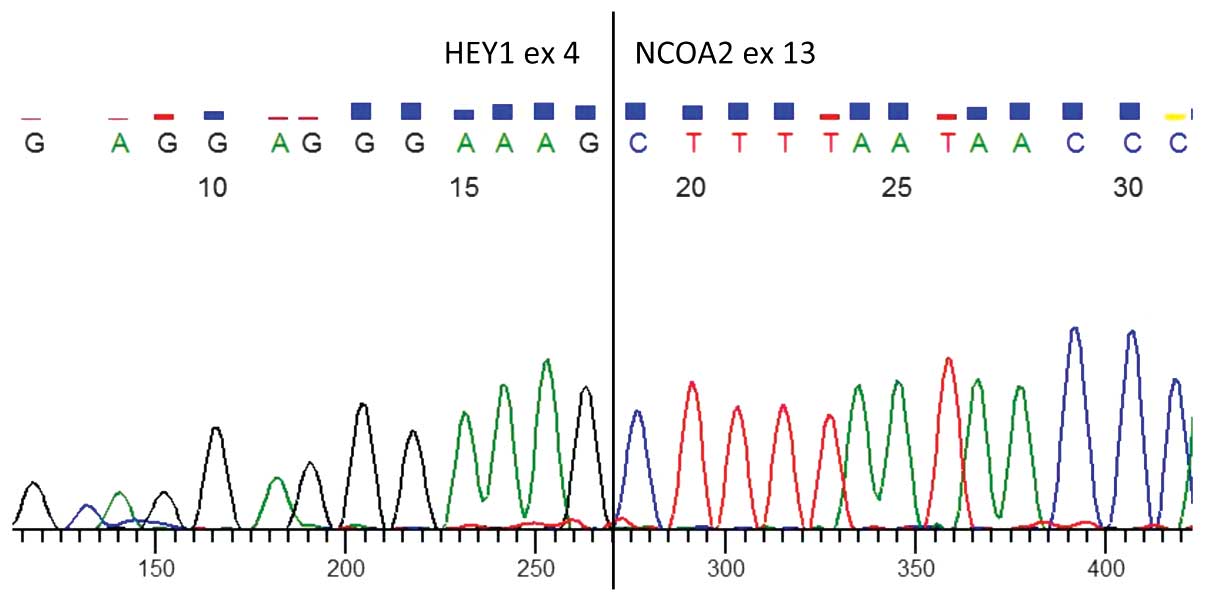
and reverse primer from exon 13 of NCOA2. Direct sequencing of the
product confirmed the presence of a HEY1-NCOA2 chimeric transcript
with a junction between HEY1 exon 4 and NCOA2 exon 13 (Figs. 2 and 3).
Discussion
In the current study, the HEY1-NCOA2 fusion gene was
detected in a paediatric case of intraspinal MCS. These findings
validate those of a recently published primary report regarding
this gene fusion in MCS at an intraspinal location (19).
MCS, as observed in the current case, is an
extremely rare tumour that is morphologically characterized by a
biphasic pattern of small, round, undifferentiated hyperchromatic
cells with islands of cartilage with a varying degree of hyaline
differentiation. Approximately 70% of all MCS cases occur in the
bone, with the remaining detected in extraskeletal locations, as
observed in this case. In contrast to classical chondrosarcomas,
MCS is an aggressive, fast-growing tumour that frequently
metastasises and has the ability to remain dormant for long periods
of time. Furthermore, as seen in this case, MCS tends to affect
children and young adults, in contrast to classical chondrosarcomas
that mostly affect patients over 50 years of age. MCS accounts for
<1% of all sarcomas (20) and
represents only 2–10% of all chondrosarcomas (2). The tumour preferentially metastasises
to the lung, lymph nodes and other bones (6,7). To
date, with the present case included, only 15 cases of intraspinal
(meningeal) MCS have been published (6,7,21–32),
and seven of these occurred in children (7,21,24,27,30–32).
These tumours arise most frequently in the mid-level of the spine,
specifically, the lower thoracic and upper lumbar region; although,
in certain cases, tumours have been identified in the cervical
spine (26,33) as well as the sacral part of the
spine (22).
As expected, symptoms associated with these tumours
reflect their compressive effects on the specific neuroanatomical
structure. As was the condition of the present case, the clinical
findings of MCS are often subtle and non-specific. Typically the
patient presents with focal back pain, stiffness and, on occasion,
sensory-motor signs of spinal cord compression, such as weakness.
The duration of symptoms also differs considerably, ranging from
weeks to many years, often leading to late investigation and
diagnosis. However, in the majority of reported cases, the initial
symptom is pain, probably as a consequence of local swelling,
similar to the current report (1,34).
In accordance with the standard investigative
procedures of bone tumours, clinical, radiological and pathological
examinations are necessary to obtain a correct diagnosis.
Radiological findings of MCS typically comprise an osteolytic
diffusely demarcated lesion with punctate calcifications. On plain
radiographs, these lesions exhibit radiolucent areas with matrix
calcification, such as arcs and rings (1,7,35).
However, no specific MRI findings to distinguish mesenchymal
chondrosarcomas from ordinary types of chondrosarcomas have been
established thus far.
The rarity of the tumour, in combination with
different origins, such as bone, soft tissue, brain, meninges and
spinal tissue (25,33,36),
prevents conclusive studies. Consequently, the underlying tumour
mechanisms are therefore poorly understood (2), and there is no general agreement with
regard to the best choice of therapy (3). Primary treatment is based on surgery,
although some centres have used adjuvant radiotherapy as well as
chemotherapy (9). The effectiveness
of adjuvant chemo- and/or radiotherapy in conjunction with surgery
is not well-defined (5,20,37–39).
Unsurprisingly, it is concluded that wide surgical resection
margins improve the survival outcome (10). Earlier studies have additionally
suggested that survival is affected negatively and positively by
specific factors, such as proliferation rate (12) and tumour origin in bone (10), respectively.
The HEY1-NCOA2 gene fusion detected in the present
study has, to date, only been detected in MCS, but has been absent
in all types of chondrosarcoma that have been investigated
(19). This finding is of clinical
as well as scientific value, since identification of a specific
molecular marker should effectively distinguish MCS from other
types of morphologically similar sarcomas, and further provide a
key to the resolution of pathogenesis, since NCOA2 interacts with
specific ligand-bound nuclear receptors that facilitate chromatin
remodelling and transcription of nuclear receptor target genes
(40). NCOA2 is included in the
nuclear receptor transcriptional co activator family (41). Notably, NCOA2 has been detected as a
fusion partner in numerous other malignancies, including acute
myeloid leukaemia (MYST3-NCOA2) (42), other types of acute leukaemia
(ETV6-NCOA2) (43), subtypes of
alveolar rhabdomyosarcoma (PAX3-NCOA2) (44) and, recently, benign soft tissue
angiofibromas (AHHR-NCOA2) (45).
These examples demonstrate that despite divergent clinical
behaviour and outcomes between different diagnoses, the tumours
commonly contain NCOA2 as a fusion partner. Further studies are
required to provide evidence of how these genes are involved in the
pathogenesis of neoplastic disorders. Furthermore, the mechanisms
underlying the high specificity of the HEY1-NCOA2 gene fusion for
MCS and its potential utility in the development of new therapeutic
approaches remain to be established.
Acknowledgements
The study was supported by the Lions Cancer
Foundation, Umeå, and the Faculty of Medicine, Umeå University
(Umeå, Sweden). We are additionally grateful to medical laboratory
scientist Miss Carina Karlsson.
References
|
1
|
Lichtenstein L and Bernstein D: Unusual
benign and malignant chondroid tumors of bone. A survey of some
mesenchymal cartilage tumors and malignant chondroblastic tumors,
including a few multicentric ones, as well as many atypical benign
chondroblastomas and chondromyxoid fibromas. Cancer. 12:1142–1157.
1959.
|
|
2
|
Huvos AG, Rosen G, Dabska M and Marcove
RC: Mesenchymal chondrosarcoma. A clinicopathologic analysis of 35
patients with emphasis on treatment. Cancer. 51:1230–1237.
1983.
|
|
3
|
Nakashima Y, Unni KK, Shives TC, Swee RG
and Dahlin DC: Mesenchymal chondrosarcoma of bone and soft tissue.
A review of 111 cases. Cancer. 57:2444–2453. 1986.
|
|
4
|
Salvador AH, Beabout JW and Dahlin DC:
Mesenchymal chondrosarcoma - observations on 30 new cases. Cancer.
28:605–615. 1971.
|
|
5
|
Bertoni F, Picci P, Bacchini P, et al:
Mesenchymal chondrosarcoma of bone and soft tissues. Cancer.
52:533–541. 1983.
|
|
6
|
Nguyen BD, Daffner RH, Dash N, Rothfus WE,
Nathan G and Toca AR Jr: Case report 790. Mesenchymal
chondrosarcoma of the sacrum. Skeletal Radiol. 22:362–366.
1993.
|
|
7
|
Kruse R, Simon RG, Stanton R, Grissom LE
and Conard K: Mesenchymal chondrosarcoma of the cervical spine in a
child. Am J Orthop (Belle Mead NJ). 26:279–282. 1997.
|
|
8
|
Dowling EA: Mesenchymal chondrosarcoma. J
Bone Joint Surg Am. 46:747–754. 1964.
|
|
9
|
Dantonello TM, Int-Veen C, Leuschner I, et
al: Mesenchymal chondrosarcoma of soft tissues and bone in
children, adolescents, and young adults: experiences of the CWS and
COSS study groups. Cancer. 112:2424–2431. 2008.
|
|
10
|
Cesari M, Bertoni F, Bacchini P, Mercuri
M, Palmerini E and Ferrari S: Mesenchymal chondrosarcoma. An
analysis of patients treated at a single institution. Tumori.
93:423–427. 2007.
|
|
11
|
World Health Organisation. Classification
of tumours. Pathology and Genetics of Tumours of Soft Tissue and
Bone. Fletcher CD, Unni KK and Mertens F: IARC Press; Lyon: pp.
12–367. 2002
|
|
12
|
Nussbeck W, Neureiter D, Söder S, Inwards
C and Aigner T: Mesenchymal chondrosarcoma: an immunohistochemical
study of 10 cases examining prognostic significance of
proliferative activity and cellular differentiation. Pathology.
36:230–233. 2004.
|
|
13
|
Turc-Carel C, Philip I, Berger MP, Philip
T and Lenoir GM: Chromosome study of Ewing’s sarcoma (ES) cell
lines. Consistency of a reciprocal translocation t(11;22)(q24;q12).
Cancer Genet Cytogenet. 12:1–19. 1984.
|
|
14
|
Turc-Carel C, Aurias A, Mugneret F, et al:
Chromosomes in Ewing’s sarcoma. I An evaluation of 85 cases of
remarkable consistency of t(11;22)(q24;q12). Cancer Genet
Cytogenet. 32:229–238. 1988.
|
|
15
|
Lessnick SL, Dacwag CS and Golub TR: The
Ewing’s sarcoma oncoprotein EWS/FL indices a p53-dependent growth
arrest in primary human fibroblasts. Cancer Cell. 1:393–401.
1999.
|
|
16
|
Le Deley MC, Delattre O, Schaefer KL, et
al: Impact of EWS-ETS fusion type on disease progression in Ewing’s
sarcoma/peripheral primitive neuroectodermal tumor: prospective
results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin
Oncol. 28:1982–1988. 2010.
|
|
17
|
Sainati L, Scapinello A, Montaldi A, et
al: A mesenchymal chondrosarcoma of a child with the reciprocal
translocation (11;22)(q24;q12). Cancer Genet Cytogenet. 71:144–147.
1993.
|
|
18
|
Gatter KM, Olson S, Lawce H and Rader AE:
Trisomy 8 as the sole cytogenetic abnormality in a case of
extraskeletal mesenchymal chondrosarcoma. Cancer Genet Cytogenet.
159:151–154. 2005.
|
|
19
|
Wang L, Motoi T, Khanin R, et al:
Identification of a novel, recurrent HEY1-NCOA2 fusion in
mesenchymal chondrosarcoma based on a genome-wide screen of
exon-level expression data. Genes Chromosomes Cancer. 51:127–139.
2012.
|
|
20
|
Dabska M and Huvos AG: Mesenchymal
chondrosarcoma in the young. Virchows Arch A Pathol Anat
Histopathol. 399:89–104. 1983.
|
|
21
|
Daita G, Abe H, Itoh T, Nakagawa T, Tsuru
M and Hirama M: A case of mesenchymal chondrosarcoma originating
from the spinal dura (author’s transl). No Shinkei Geka. 7:785–790.
1979.(In Japanese).
|
|
22
|
Di Lorenzo N, Palatinsky E, Artico M and
Palma L: Dural mesenchymal chondrosarcoma of the lumbar spine. Case
report. Surg Neurol. 31:470–472. 1989.
|
|
23
|
Harsh GR IV and Wilson CB: Central nervous
system mesenchymal chondrosarcoma. Case report. J Neurosurg.
61:375–381. 1984.
|
|
24
|
Huckabee RE: Meningeal mesenchymal
chondrosarcoma of the spine: a case report. J Magn Reson Imaging.
1:93–95. 1991.
|
|
25
|
Lee ST, Lui TN and Tsai MD: Primary
intraspinal dura mesenchymal chondrosarcoma. Surg Neurol. 31:54–57.
1989.
|
|
26
|
Ranjan A, Chacko G, Joseph T and Chandi
SM: Intraspinal mesenchymal chondrosarcoma. Case report. J
Neurosurg. 80:928–930. 1994.
|
|
27
|
Scheithauer BW and Rubinstein LJ:
Meningeal mesenchymal chondrosarcoma: report of 8 cases with review
of the literature. Cancer. 42:2744–2752. 1978.
|
|
28
|
Zucker DK and Horoupian DS: Dural
mesenchymal chondrosarcoma. Case report. J Neurosurg. 48:829–833.
1978.
|
|
29
|
Platania N, Nicoletti G, Lanzafame S and
Albanese V: Spinal meningeal mesenchymal chondrosarcoma. Report of
a new case and review of the literature. J Neurosurg Sci.
47:107–110. 2003.
|
|
30
|
Chen SH, Wang HS, Jaing TH, Hsueh C, Lo WC
and Tseng CK: Primary intraspinal mesenchymal chondrosarcoma:
report of one case. Acta Paediatr Taiwan. 46:308–310. 2005.
|
|
31
|
Reif J and Graf N: Intraspinal mesenchymal
chondrosarcoma in a three-year-old boy. Neurosurg Rev. 10:311–314.
1987.
|
|
32
|
Chan HS, Turner-Gomes SO, Chuang SH, et
al: A rare cause of spinal cord compression in childhood from
intraspinal mesenchymal chondrosarcoma. A report of two cases and
review of the literature. Neuroradiology. 26:323–327. 1984.
|
|
33
|
Rushing EJ, Armonda RA, Ansari Q and Mena
H: Mesenchymal chondrosarcoma: a clinicopathologic and flow
cytometric study of 13 cases presenting in the central nervous
system. Cancer. 77:1884–1891. 1996.
|
|
34
|
Zibis AH, Wade Shrader M and Segal LS:
Case report: Mesenchymal chondrosarcoma of the lumbar spine in a
child. Clin Orthop Relat Res. 468:2288–2294. 2010.
|
|
35
|
Theodorou DJ, Theodorou SJ, Xenakis T,
Demou S, Agnantis N and Soucacos PN: Mesenchymal chondrosarcoma of
soft tissues of the calf. Am J Orthop (Belle Mead NJ). 30:329–332.
2001.
|
|
36
|
Huvos AG and Marcove RC: Chondrosarcoma in
the young. A clinicopathologic analysis of 79 patients younger than
21 years of age. Am J Surg Pathol. 11:930–942. 1987.
|
|
37
|
Harwood AR, Krajbich JI and Fornasier VL:
Mesenchymal chondrosarcoma: a report of 17 cases. Clin Orthop Relat
Res. 158:144–148. 1981.
|
|
38
|
Guccion JG, Font RL, Enzinger FM and
Zimmerman LE: Extraskeletal mesenchymal chondrosarcoma. Arch
Pathol. 95:336–340. 1973.
|
|
39
|
Tuncer S, Kebudi R, Peksayar G, et al:
Congenital mesenchymal chondrosarcoma of the orbit: case report and
review of the literature. Ophthalmology. 111:1016–1022. 2004.
|
|
40
|
Voegel JJ, Heine MJ, Tini M, Vivat V,
Chambon P and Gronemeyer H: The coactivator TIF2 contains three
nuclear receptor-binding motifs and mediates transactivation
through CBP binding-dependent and -independent pathways. Embo J.
17:507–519. 1998.
|
|
41
|
Xu J and Li Q: Review of the in vivo
functions of the p160 steroid receptor coactivator family. Mol
Endocrinol. 17:1681–1692. 2003.
|
|
42
|
Carapeti M, Aguiar RC, Goldman JM and
Cross NC: A novel fusion between MOZ and the nuclear receptor
coactivator TIF2 in acute myeloid leukemia. Blood. 91:3127–3133.
1998.
|
|
43
|
Strehl S, Nebral K, Konig M, et al:
ETV6-NCOA2: a novel fusion gene in acute leukemia associated with
coexpression of T-lymphoid and myeloid markers and frequent NOTCH1
mutations. Clin Cancer Res. 14:977–983. 2008.
|
|
44
|
Sumegi J, Streblow R, Frayer RW, et al:
Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma
without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear
receptor transcriptional coactivator family. Genes Chromosomes
Cancer. 49:224–236. 2010.
|
|
45
|
Jin Y, Möller E, Nord KH, et al: Fusion of
the AHRR and NCOA2 genes through a recurrent translocation
t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation
of aryl hydrocarbon receptor target genes. Genes Chromosomes
Cancer. 51:510–520. 2012.
|