Introduction
Giant cell tumors (GCTs) are considered to be a
locally aggressive benign tumors, also known as osteoclastoma,
which typically occur in the epiphyses of long bones, particularly
the distal femur, proximal tibia, distal radius and proximal
humerus. GCT rarely manifests in the skull, accounting for <1%
of all GCTs of the bone, primarily involving the sphenoid and
temporal bones in the middle of the cranial fossa (1–3). At
present, the majority of studies regarding GCT of the skull are
case reports, and the bones involved include temporal bone,
petrosal bone, sphenoid and occipital bone. Primary GCT of the
clivus is extremely rare. Due to the small number of skull GCTs
reported in the literature, standard treatments remain unclear, and
the efficacy of surgery as well as adjuvant therapies remains
undefined. The current study presents a case of GCT in the clivus
presenting with abducens nerve and trigeminal nerve involvement
concurrently in a 22-year-old male, who was treated successfully
with minimally invasive surgery, adjuvant radiotherapy and
intravenous bisphosphonates, and the literature regarding
diagnosis, treatment and prognosis has been reviewed. Written
informed consent was obtained from the patient’s family.
Case report
A 22-year-old male with a six month history of dull
frontal headache did not receive any medical treatment or
examination as the symptoms were tolerable. However, three days
prior to admission, the headache worsened, and was localized to the
right side. The patient developed secondary diplopia and facial
numbness in the right maxilla area. On ophthalmological
examination, the diplopia secondary to the left abducens nerve (CN
VI) palsy was observed in the left eye. Examination of the cranial
nerves revealed facial paresthesia along the distribution of
maxillary (V2) divisions of the right trigeminal nerve (CN V). No
abnormalities in vision, visual field, corneal reflexes, hearing or
the power of masseters were identified and no papilledema was
observed. The remaining motor and sensory neurological
examinations, including cerebellar tests, were normal with full
cooperation and orientation. Endoscopic nasal examination revealed
a soft, friable mass, which bled when palpated in the posterior
wall of the nasopharynx top. The laboratory evaluations including,
complete blood count, biochemistry, analysis of tumor markers,
thyroid and pituitary function tests and endocrinology examinations
were normal, and the patient’s medical history was noncontributory.
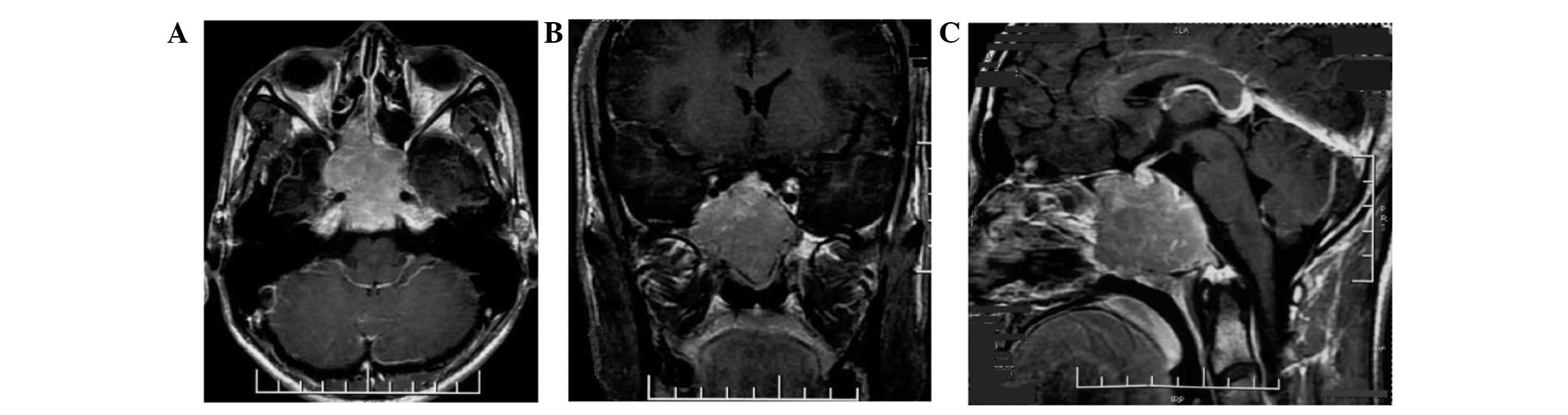
Magnetic resonance imaging (MRI) of the brain demonstrated an
extensive soft-tissue density mass with an irregular shape and a
clear boundary measuring 4.0×4.68×3.7 cm, involving the clivus,
surrounding the cavernous sinuses on both sides and compressing the
front of optic chiasm in the sphenoid sinus area of middle fossa.
The posterior wall of the nasopharynx top was not involved. The
tumor tissue was isointense on T1-weighted imaging (WI), T2WI and
fluid-attenuated inversion recovery, and moderate homogenous
enhancement was identified on the post contrast scan (Fig. 1). Due to the symptoms of the present
illness and MRI imaging, chordoma and malignant tumor in clivus
could not be excluded.
The tumor was removed using an endoscopic transnasal
transsphenoidal method under general anesthesia. Intraoperative
findings revealed a gray, soft, friable, hypervascular mass arising
from the clivus and involving the sphenoid and posterior ethmoid
sinuses. Consequently, almost total resection included the tumor
together with the slopes and bone surrounding the optic canal and
all cranial nerves were preserved. Postoperatively, the patient
exhibited transient diabetes insipidus.
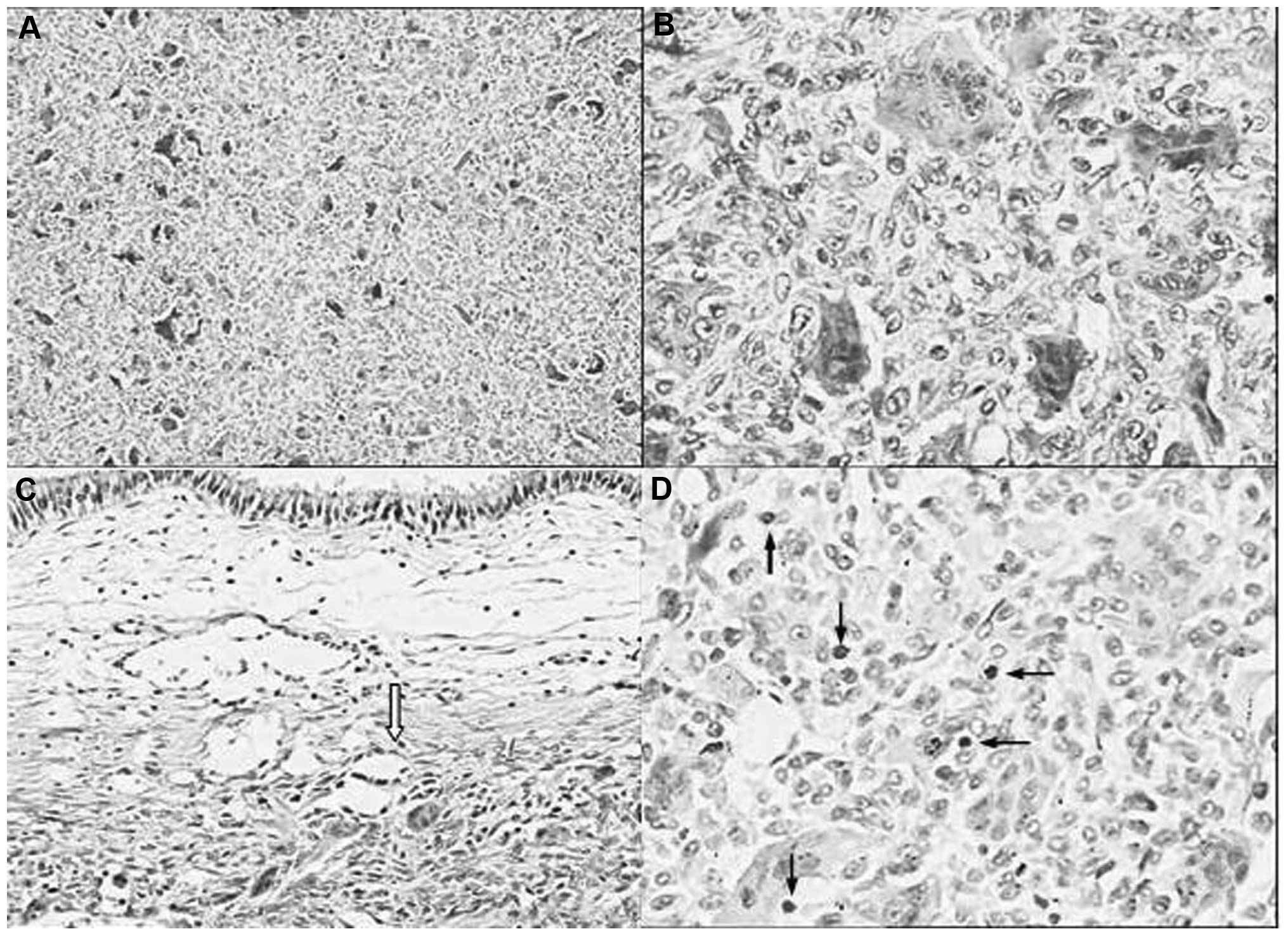
From the postoperative histopathology, the patient
was diagnosed with a giant cell tumor. The histopathology revealed
the tumor was composed of flaky oval or spindle-shaped mononuclear
stromal cells and evenly distributed osteoclast-like multinucleated
giant cells. The giant cells contained a variable number of nuclei
with an median of 25 (range, 10–30). The nuclear features of
stromal cells were similar to multinucleated giant cells, with open
chromatin, one to two nucleoli and numerous mitotic figures (≤15
per 10 high-power fields). No pathological mitosis was identified
(Fig. 3).
Postoperatively, the patient was administered three
courses of intravenous zoledronate (4 mg, once a month) and
radiotherapy to reduce the local recurrence caused by subtotal
resection of the tumor. The patient received three-dimensional
conformal radiotherapy using five coplanar fields and two
non-coplanar fields to deliver a total dose of 45 Gy in 25
fractions over five weeks. The dose volume histogram revealed that
92% of the planning treatment volume was receiving 100% of the
dose. All critical structures received doses within their limits of
tolerance. The patient completed therapy without any significant
acute toxicity. Complete regression of the tumor was later
confirmed on MRI following treatment.
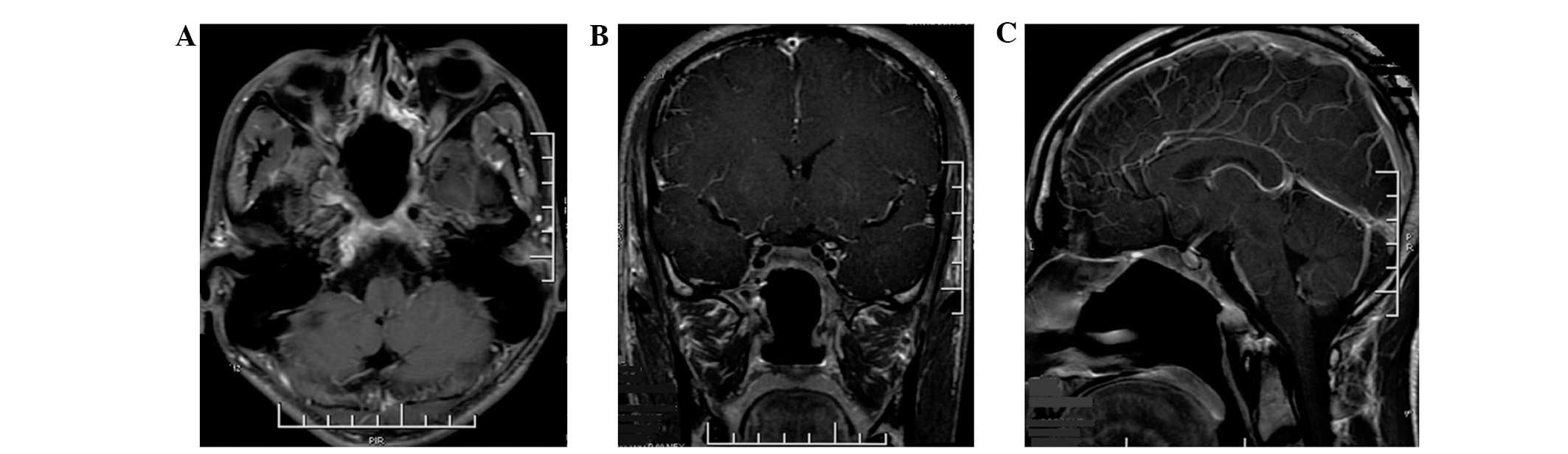
Follow-up has been performed for two years, the
patient is clinically asymptomatic and no evidence of recurrence or
metastases has been identified by computed tomography (CT) and MRI
examination (Fig. 2).
Discussion
GCTs are generally considered histologically benign;
however, they may exhibits locally aggressive behavior with a high
rate of local recurrence of up to 60% if treated purely by
intralesional curettage. In adiditon, GCTs exhibit the potential
for distant metastasis, mostly commonly to the lung, which occurs
in 4% of patients with GCT (7). The
incidence of GCT is low, accounting for only ~4–5% of primary
tumors of the skeleton; however, it is relatively more common in
Asian populations, accounting for 14.2%of primary tumors of the
skeleton in China, and it occurs more frequently in females than in
males, between the second and fourth decades of life following
skeletal maturation (8). GCTs most
frequently occur in the metaphyses of long bones, but rarely in the
skull, accounting for <1% of bone GCTs, where it is usually
located in sphenoid and temporal bone. However, in the present
case, the tumor primarily arose from the clivus with sellar
extension. During the past decade, only five cases of primary
clival GCT have been reported (Table
I).
 | Table ISummary of giant cell tumor of clivus
reported in the English literature. |
Table I
Summary of giant cell tumor of clivus
reported in the English literature.
| No. | Author (ref.) | Patient age (years);
Gender | Location | Presentations | Therapy | Histology | Recurrence | Patient status;
Follow-up |
|---|
| 1 | Zorlu et al
(4) | 14; Female | Sphenoid, clivus | Headache,
diplopia | Neuro-navigation
guided transsphenoidal Surgery and radiotherapy (60 Gy) on
recurrence | Malignant GCT | Recurrent both after
surgery and after radiotherapy | AWD; 2 years |
| 2 | Gupta et al
(12) | 17; Female | Clivus | Diplopia, amenorrhea,
decreased vision, headache (bilateral CN6 palsy and left CN5 palsy
partially) | Surgery via LeFort I
osteotomyand and radiotherapy (45 Gy) | Malignant GCT | No | ANED; 2 years |
| 3 | Sasagawa et al
(5) | 26; Male | Clivus | Headache, diplopia
(right CN6 palsy) | Transsphenoidal
surgery and radiotherapy (50 Gy); Transsphenoidal surgery,
chemotherapy and artery embolization after recurrence | GCT, osteosarcoma
after recurrence | Malignant
transformation and lung metastasis | DWD; 10 years |
| 4 | Roy et al
(3) | 19; Male | Clivus | Headache, forehead
and cheek numbness (right CN5 palsy) | Surgery via right
trans-maxillary approach and radiotherapy (45 Gy) | GCT | No | ANED; 18 months |
| 5 | Iacoangeli et
al (6) | 31; Male | Clivus | Headache, diplopia
(right CN6 palsy) | Surgery via
endoscopic extended endonasal approach (EEA) | GCT | No | ANED; 6 years |
Clinical manifestations are usually in accordance
with the site of the tumor. Skull-base GCTs generally present with
headache, decreased vision, visual field defect, diplopia,
ophthalmoplegia, deafness, endocrinopathy and dysfunction of
cranial nerves, most commonly the sixth followed by the third
cranial nerve (6). However, our
patient developed diplopia and facial numbness, which indicated the
involvement of the sixth and the fifth cranial nerves. Similar
cases with sixth and fifth cranial nerve involvement concurrently
have rarely been reported (3,9).
X-ray and CT scan of skull GCTs frequently
demonstrate an expansive and occasional lytic bone lesions usually
without the classical ‘soap bubble’ appearance. MRI clearly
demonstrates the soft-tissue extension and association with the
surrounding structures. On MRI, GCTs are usually hypointense or
isointense on T1-weighted images (WI) and T2WI with contrast
enhancement (10,11). A similar pattern was observed in the
present case. The major radiological differential diagnoses include
chordoma, giant-cell reparative granuloma, aneurysmal bone cyst,
fibrous dysplasia, ‘brown tumor’ of hyperparathyroidism,
eosinophilic granuloma and plasmacytoma. Imaging examination alone
is insufficient to differentiate these lesions and, thus, the final
diagnosis is dependent on histopathology.
GCTs of the bone originate from the primary
mesenchymal stromal cells in the connective tissue of the bone
marrow, which expresses the receptor activator of NF-κB ligand that
stimulates osteoclast maturation from mononuclear precursors.
Histologically, GCTs are primarily composed of mononuclear stromal
cells and giant cells. However, histogenesis is controversial as
the terms GCT and osetoclastoma imply that the giant cells are
responsible for the proliferative capacity of the tumor, however,
the mononuclear cells present the true neoplastic component and the
multinucleated giant cells exhibit an osetoclast-like phenotype and
express histocytic lineage markers. The mononuclear cell presents
the true neoplastic component while the multinucleated giant cells
exhibit an osteoclast-like phenotype and express histocytic lineage
markers (12). Multiple cytogenetic
abnormalities associated with GCTs have been reported, in which
telomere adhesion was the most frequent chromosomal aberration
(75%) (13). Cellular morphology is
sufficient for the diagnosis of GCT, and immunochemistry is not
essential. However, the lineage of these cells may be determined
using histiocytic marker CD68 immunostain (12).
The clinical behavior of GCT is unpredictable and,
thus, treatment remains controversial. Radical surgical extirpation
is the treatment of choice for cranial GCT, which requires complete
removal of the diseased bone. However, this may not be possible due
to anatomical location or the involvement of vital structures, as
observed in the present case, and thus the patient was treated
using a minimally intralesional approach. Therefore, the recurrence
rate is very high, and the use of adjuvant therapy is invaluable
(14,15).
GCTs were previously considered to be
radio-resistant with a potential for sarcomatous transformation
following radiotherapy. However, along with the development of
modern megavoltage irradiation and precise image guided system, the
tumor control rate has significantly improved and the frequency of
malignant transformation has reduced. Therefore, radiotherapy is
recommended as a postoperative adjunctive therapy particularly for
incomplete resection in skull base, with a dose of 45–50 Gy in
order to gain a long-term healing (16). The role of chemotherapy remains
unclear and controversial. A small number of studies have
demonstrated that chemotherapy results in good control of primary
or recurrent GCT of the skull base (17–19).
At present no effective chemotherapeutic agents have been
identified for the treatment of this tumor. We suggest that
systemic chemotherapy be considered if local control fails
following radiotherapy or distal metastases are identified. Studies
have indicated that topical or systemic use of bisphosphonates may
present a novel adjuvant therapy for GCT by inducing apoptosis of
stromal tumor cells and stimulating osteogenic differentiation of
the remaining tumor stromal cells following surgery (20–22).
Radiographical and histological grading systems do
not predict clinical outcome; however, the extent of surgical
resection has been shown to affect prognosis. The majority of
recurrences occur within the first two years following treatment,
although late recurrences have also been reported and, thus,
long-term surveillance is recommended (23).
Giant cell tumors are generally benign, locally
aggressive lesions with a potential to metastasize. Primary GCTs of
the clivus are extremely rare and only five cases have been
reported during the past decade. Imaging examination alone is
insufficient for the diagnosis of GCT in the skull-base, and the
final diagnosis is dependent on histopathology. Surgical
extirpation is the standard treatment for skull-base GCT, and
adjuvant radiation must be applied in all cases due to the high
rate of local recurrence and since complete resection cannot be
achieved. Bisphosphonate administration is also recommended. The
present case indicated that the use of subtotal excision via
minimally invasive surgery following the administration of
intravenous bisphosphonate and adjuvant radiotherapy (45 Gy)
results in excellent tumor control after a period of two years’
follow-up. Recurrences usually occur within the first two years
following treatment, however, long-term surveillance is
proposed.
References
|
1
|
Pai SB, Lalitha RM, Prasad K, et al: Giant
cell tumor of the temporal bone - a case report. BMC Ear Nose
Throat Disord. 5:8–15. 2005.
|
|
2
|
Leonard J, Gökden M, Kyriakos M, et al:
Malignant giant-cell tumor of the parietal bone: case report and
review of the literature. Neurosurg. 48:424–429. 2001.
|
|
3
|
Roy S, Joshi NP, Sigamani E, et al: Clival
giant cell tumor presenting with isolated trigeminal nerve
involvement. Eur Arch Otorhinolaryngol. 270:1167–1171. 2013.
|
|
4
|
Zorlu F, Selek U, Soylemezoglu F and Oge
K: Malignant giant cell tumor of the skull base originating from
clivus and sphenoid bone. J Neurooncol. 76:149–152. 2006.
|
|
5
|
Sasagawa Y, Tachibana O, Shiraga S, et al:
Secondary malignant giant cell tumor of the clivus: case report.
Clin Neurol Neurosurg. 114:786–788. 2012.
|
|
6
|
Iacoangeli M, Di Rienzo A, Re M, et al:
Endoscopic endonasal approach for the treatment of a large clival
giant cell tumor complicated by an intraoperative internal carotid
artery rupture. Cancer Manag Res. 5:21–24. 2013.
|
|
7
|
Carrasco CH and Murray JA: Giant cell
tumors. Orthop Clin North Am. 20:395–405. 1989.
|
|
8
|
Wang Y, Honda K, Suzuki S, et al: Giant
cell tumor at the lateral skull base. Am J Otolaryngol. 27:64–67.
2006.
|
|
9
|
Weber AL, Hug EB, Muenter MW and Curtin
HD: Giant-cell tumors of the sphenoid bone in four children:
radiological, clinical, and pathological findings. Skull Base Surg.
7:163–173. 1997.
|
|
10
|
Sharma RR, Mahapatra AK, Pawar SJ, et al:
Craniospinal giant cell tumors: clinicoradiological analysis in a
series of 11 cases. J Clin Neurosci. 9:41–50. 2002.
|
|
11
|
Huang PH, Lee CC, Chang PY, et al: Giant
cell tumor of the sphenoid bone occurring during pregnancy:
successful tumor extirpation via endoscopic transnasal
transsphenoidal surgery. Clin Neurol Neurosurg. 115:222–226.
2013.
|
|
12
|
Zheng MH, Robbins P, Xu J, et al: The
histogenesis of giant cell tumour of bone: a model of interaction
between neoplastic cells and osteoclasts. Histol Histopathol.
16:297–307. 2001.
|
|
13
|
Company MM and Ramos R: Giant cell tumor
of the sphenoid. Arch Neurol. 66:134–135. 2009.
|
|
14
|
Campanacci M, Baldini N, Boriani S, et al:
Giant-cell tumor of bone. J Bone Joint Surg Am. 69:106–114.
1987.
|
|
15
|
Gupta R, Mohindra S, Mahore A, et al:
Giant cell tumor of the clivus. Br J Neurosurg. 22:447–449.
2008.
|
|
16
|
Ruka W, Rutkowski P, Morysinski T, et al:
The megavoltage radiation therapy in treatment of patients with
advanced or difficult giant cell tumors of bone. Int J Radiat Oncol
Biol Phys. 78:494–498. 2010.
|
|
17
|
Yamamoto M, Fukushima T, Sakamoto S, et
al: Giant cell tumor of the sphenoid bone: long-term follow-up of
two cases after chemotherapy. Surgical Neurology. 49:547–552.
1998.
|
|
18
|
Moon JC, Kim SR, Chung MJ and Lee YC:
Multiple pulmonary metastases from giant cell tumor of a hand. Am J
Med Sci. 343:171–173. 2012.
|
|
19
|
Shirzadi A, Drazin D, Bannykh S and
Danielpour M: Giant cell tumor of the odontoid in an adolescent
male: radiation, chemotherapy, and resection for recurrence with
10-year follow-up. J Neurosurg Pediatr. 8:367–371. 2011.
|
|
20
|
Chang SS, Suratwala SJ, Jung KM, et al:
Bisphosphonates may reduce recurrence in giant cell tumor by
inducing apoptosis. Clin Orthop Relat Res. 426:103–109. 2004.
|
|
21
|
Cheng YY, Huang L, Lee KM, et al:
Bisphosphonates induce apoptosis of stromal tumor cells in giant
cell tumor of bone. Calcif Tissue Int. 75:71–77. 2004.
|
|
22
|
Yang T, Zheng XF, Li M, et al: Stimulation
of osteogenic differentiation in stromal cells of giant cell tumour
of bone by zoledronic Acid. Asian Pac J Cancer Prev. 14:5379–5383.
2013.
|
|
23
|
Puri A and Agarwal M: Treatment of giant
cell tumor of bone: current concepts. Indian J Orthop. 41:101–108.
2007.
|