Introduction
Double-stranded RNA-dependent protein kinase (PKR,
also known as EIF2AK2) was originally identified as a
first-response protein, which induces cell defense responses upon
viral infection (1). Briefly,
double-stranded RNA, usually produced during virus replication by
viral RNA polymerases, binds protein PKR, facilitates the
homo-dimerization and auto-phosphorylation of PKR at Thr451 and
Thr446, and thereby activates PKR (2,3). PKR
has also been found to be activated by other stress signals,
therefore serving as a signaling hub of the proinflammatory
response to stimuli including bacterial lipopolysaccharide, tumor
necrosis factor α and interleukin 1 (4). In the canonical PKR signaling pathway,
PKR serves as a eukaryotic initiation factor 2α (eIF-2α) kinase,
which promotes the phosphorylation of eIF-2α at Ser51 (3). Phosphorylated eIF-2α inhibits the
initiation of translation, resulting in the suppression of general
protein synthesis and therefore suppression of cell growth and
induction of cellular apoptosis, in numerous types of eukaryotic
cell (3,5). Therefore, PKR has been previously
suggested to be a tumor suppressor due to its potential for
inhibiting cell growth and inducing apoptosis (6–8). For
example, in liver cancer cells, PKR-activating agents, such as
interferon and radicicol, were shown to enhance the apoptotic
effect of the transcription factor E2F1, a process proposed to be
mediated by transcriptionally upregulated PKR expression (8). However, these effects were marginal,
and there remains a lack of direct evidence and detailed mechanisms
to show the exact role of PKR in liver cancer development.
Multiple studies have observed increased expression
levels and elevated activity of PKR in hepatitis C virus
(HCV)-related and -unrelated hepatocellular carcinoma (9–11), as
well as in several other cancer cell types, for example, human
breast cancer cells (12) and
melanoma cells (13,14). Elevated PKR expression levels and
activity may be a tumor marker, and may contribute to the
proliferation of tumor cells and tumor development (9,12–14).
Studies have also suggested that PKR may suppress apoptosis by
activating the nuclear factor κB (NF-κB) signaling pathway
(15) and the corresponding target
gene, B-cell lymphoma 2 (Bcl2), an antiapoptotic protein (16). However, this remains indirect
evidence, although this does suggest a potential tumorigenic role
of PKR in hepatocellular carcinoma. In addition, although the
positive effect of PKR on the antiapoptotic pathway through NF-κB
and Bcl2 has been demonstrated, whether PKR exerts any effect on
pro-proliferation transcriptional pathways remains unknown.
PKR has been found to activate several transcription
factors, including IRF-1, p53 and NF-κB (17,18).
PKR has also been shown to directly bind with STAT3 and regulate
STAT3 transcriptional activity, although whether PKR activates or
suppresses STAT3 remains controversial (19,20).
Elevated STAT3 activity, which depends on phosphorylation, has been
observed in primary liver cancer (21). Hepatocyte-specific STAT3-deficient
mice exhibited markedly greater resistance to hepatocellular
carcinoma and the tumor sizes were clearly smaller, suggesting that
STAT3 is crucial in promoting hepatocellular carcinoma cell
proliferation and/or survival (22).
The present study aimed to provide direct evidence
with regard to the function of PKR in liver cancer tumorigenesis
via in vivo and in vitro assays, and to describe the
detailed underlying mechanism. Furthermore this study aimed to
clarify the oncogenic role of PKR in hepatocellular carcinoma and
detail the mechanism.
Materials and methods
Patients and tissues
Tumor and adjacent normal tissue samples were
obtained from four primary liver cancer patients (two males and two
females) at the Department of Hepatobiliary Surgery, The General
Hospital of Chinese People’s Liberation Army (Beijing, China) in
2012. The patient’s ages ranged between 50 and 60 years. The four
patients were first-time diagnosed with primary liver cancer,
without Hepatitis B or C virus infection. The normal and tumor
samples were collected during the first surgical procedures
performed in 2012, subsequent to diagnosis, and were freshly
cryo-preserved in liquid nitrogen. The frozen tissue samples were
grinded using a Spex 6770 Freezer/Mill system (SPEX SamplePrep LLC,
Metuchen, NJ, USA). This study was approved by the ethics committee
of The General Hospital of Chinese People’s Liberation Army and
written informed consent was obtained from all patients.
Cell culture and reagents
HepG2 human hepatocellular carcinoma cells (American
Type Culture Collection, Manassas, VA, USA) were cultured in
Dulbecco’s modified Eagle’s Medium (cellgro®; Mediatech,
Inc., Manassas, VA, USA) with 10% fetal bovine serum (FBS; Thermo
Fisher Scientific, Rockford, IL, USA). Freshly trypsinized HepG2
cells were suspended at 1×105 cells/ml in standard HepG2
culture medium and seeded at a density of 2×104 cells
per well in standard 24-well tissue culture plates. Subsequent to
seeding, the cells were incubated at 37°C in a 90% air/10%
CO2 atmosphere, and 500 μl fresh medium was supplied
every other day to the cultures following removal of the
supernatant.
Small interfering RNA (siRNA)-mediated
RNA interference and reverse transfection
Silencer® Select Validated siRNA
targeting human PKR and STAT3 was purchased from Ambion (Austin,
TX, USA). The synthesized oligonucleotides were as follows: PKR
siRNA, sense, 5′-GGUGAAGGUAGAUCAAAGATT-3′ and anti-sense,
5′-UCUUUGAUCUACCUUCACCTT-3′; STAT3 siRNA, sense,
5′-GCACAATCTACGAAGAATCAATT-3′ and anti-sense,
5′-TTGATTCTTCGTAGATTGTTT-3′. As described previously (16), transfection of siRNA was performed
with Lipofectamine RNAiMAX transfection reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA). Scrambled non-targeting siRNA
served as a negative control. Titration of the siRNA and the
transfection reagent was performed (not shown), and the lowest
viable siRNA and transfection reagent quantities were subsequently
applied in the loss-of-function (LOF) experiments.
Lentivirus-mediated RNA interference
Short hairpin RNA (shRNA) sequences were obtained
from Public TRC Portal (Broad Institute, Cambridge, MA, USA) and
lentiviruses expressing the shRNA sequences were synthesized by
Shanghai GenePharma Co., Ltd., (Shanghai, China). Polybrene and
puromyocin were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Transfection of the HepG2 cells with lentiviral particles (Shanghai
GenePharma Co., Ltd.)was conducted as described previously
(23). Titration of the
lentiviruses was performed (not shown), and the lowest functional
quantities of the virus (MOI=5) and polybrene were subsequently
applied in the LOF experiments. The shRNA target sequences for PKR
were as follows: shPKR-1, 5′-GCTGAACTTCTTCATGTATGT-3′ and shPKR-2,
5′-GAGGCGAGAAACTAGACAAAG-3′. The shRNA target sequence for STAT3
was 5′-GCACAATCTACGAAGAATCAA-3′.
Overexpression of PKR and forward
transfection
A pCMV6-XL5-hPKR plasmid and an empty vector,
pCMV6-XL5, were purchased from Origene (Rockville, MD, USA). As
described previously (16), forward
transfection of the plasmid was performed with the Lipofectamine
2000 (Invitrogen Life Technologies) transfection reagent, following
the manufacturer’s instructions. The cells attached to the
culturing surface were washed with phosphate-buffered saline (PBS)
and the medium was replaced with 100 μl Opti-MEM®
(Invitrogen Life Technologies) with 2% fetal bovine serum.
Subsequently, 400 ng plasmid per well in a 24-well plate were mixed
with 1 μl/well Lipofectamine 2000 in Opti-MEM and, 20 min later,
the mixture was added to the cells. After 6 h of transfection, the
cells were cultured in regular medium for 24 h and subsequently
harvested.
Western blot analysis
The HepG2 cells or tissue samples were lysed as
described previously (16) with
RIPA buffer (Cell Signaling Technology, Inc., Beverly, MA, USA).
Plated cells or cryo-grinded tissue samples were incubated with
RIPA lysis buffer (Cell Signaling Technology, Inc.) for 10 min,
then collected and sonicated briefly. Next the samples were
centrifuged at 14,000 × g for 10 min in a cold microfuge (5424R;
Eppendorf, Hamburg, Germany). Total protein levels were quantified
using a bicinchoninic assay kit from Pierce Biotechnology, Inc.
(Rockford, IL, USA). Subsequently, 20–40 μg total protein was
resolved using SDS-PAGE gels (Bio-Rad, Hercules, CA, USA),
transferred to nitrocellulose membranes, and then probed with
primary antibodies (1:1,000) overnight and secondary antibody
(1:5,000) for 1 h. Biotinylated protein ladders (Cell Signaling
Technology, Inc.) were loaded onto one well of each SDS-PAGE gel
and anti-biotin antibody was employed to detect these protein
ladders on western blots. An enhanced chemiluminescence kit (Pierce
Biotechnology, Inc.) was used for the antibody detection, and
images were captured using the Molecular Imager ChemiDoc XRS system
(Bio-Rad). Monoclonal rabbit anti-human STAT3 (Tyr705), monoclonal
mouse anti-human STAT3 and polyclonal rabbit anti-human STAT3
(Ser727) antibodies were purchased from Abcam (Cambridge, MA, USA),
and monoclonal mouse anti-β-actin, polyclonal rabbit anti-PKR and
polyclonal rabbit anti-PKR (Thr451) antibodies were obtained from
Sigma-Aldrich. Horseradish peroxidase-conjugated secondary
polyclonal goat anti-rabbit and anti-mouse antibodies were
purchased from Pierce Biotechnology, Inc.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the cells and tissue
samples with an RNeasy mini kit (Qiagen, Valencia, CA, USA) and was
depleted of contaminating DNA with RNase-free DNase (Qiagen). Equal
quantities of total RNA (1 μg) were reverse-transcribed using a
High Capacity cDNA Reverse Transcription kit (Applied Biosystems,
Carlsbad, CA, USA). The first-strand cDNA served as a template. The
primers used for RT-qPCR analyses were as follows: Human PKR,
forward, 5′-ACTTTTTCCTGGCTCATCTC-3′ and reverse,
5′-ACATGCCTGTAATCCAGCTA-3′; and human GAPDH, forward,
5′-AACTTTGGTATCGTGGAAGGA-3′ and reverse,
5′-CAGTAGAGGCAGGGATGATGT-3′, and were synthesized by Invitrogen
Life Technologies. qPCR was performed as described previously
(16). The SYBR® Select Master Mix
(Applied Biosystems, Carlsbad, CA, USA) was used in 20 μl PCR
reaction systems. Ct values <30 were considered to be reliable
in the assay. The human PKR expression levels were normalized to
those of GAPDH.
Cell proliferation assay
Experiments were conducted in an xCELLigence
Real-Time Cell Analyzer (RTCA) DP system (Roche, Mannheim,
Germany). The cells were seeded in 16-well plates (4,000 cells in
150 μl medium/well; E-plate 16; Roche), according to the
manufacturer’s instructions. The cell index, which is proportional
to the number of cells attached to the culturing surface, was
recorded in real-time every 1–2 h for up to 3–4 days. For each
well, the cell index recorded 4 h after seeding served as the
baseline to subsequently obtain the cell index fold changes. The
time point of 4 h after seeding was therefore used to indicate time
point zero in Fig. 2A. The average
fold changes in the cell proliferation index were calculated from
at least four replicate experiments, and are shown as the mean ±
standard error (SE).
Cell migration assay
Cell migration experiments were conducted in the
xCELLigence RTCA DP system (Roche). The cells were suspended in
serum-free medium and seeded in the upper chambers of 16-well
CIM-Plate 16 plates (40,000 cells in 150 μl medium/well; Roche).
Regular medium with 10% FBS was added to the lower chamber of the
CIM-Plate 16. The experiment setting and plate design were similar
to those of conventional Transwell migration assays. The cell
index, which is proportional to the number of cells that migrate
through the pores of the upper chamber, was recorded in real-time
every 30 min for up to 24 h. The average cell migration index was
calculated from at least four replicate experiments, and is
presented as mean ± SE.
In vivo xenograft transplantation
assay
HepG2 cells growing exponentially in vitro
were trypsinized and harvested for tumor implantation. Male 6–8
week old nude CD-1 mice were purchased from Vital River Laboratory
Animal Technology Co., Ltd., (Beijing, China). For each condition,
five mice were injected subcutaneously in the right flank with
2×106 HepG2 cells in 0.1 ml PBS with 0.5% bovine serum
albumin (MP Biomedicals, Santa Ana, CA, USA). When tumors became
visible, the tumor volume was monitored every three days using
caliper measurements and was calculated by the following formula:
Tumor volume (mm3) = tumor length (mm) × tumor width
(mm)2 / 2. At 27 days after injection, the animals were
sacrificed using CO2, and the xenografted tumors were
isolated and weighed.
Statistical analysis
All experiments were performed at least three times
and representative results are shown. Data are presented as the
mean ± SD for the indicated number of experiments, unless specified
otherwise. One-way analysis of variance with Student’s t-test was
used to evaluate statistical significances amongst the different
treatment groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
PKR is upregulated in hepatocellular
carcinoma tumor tissue samples
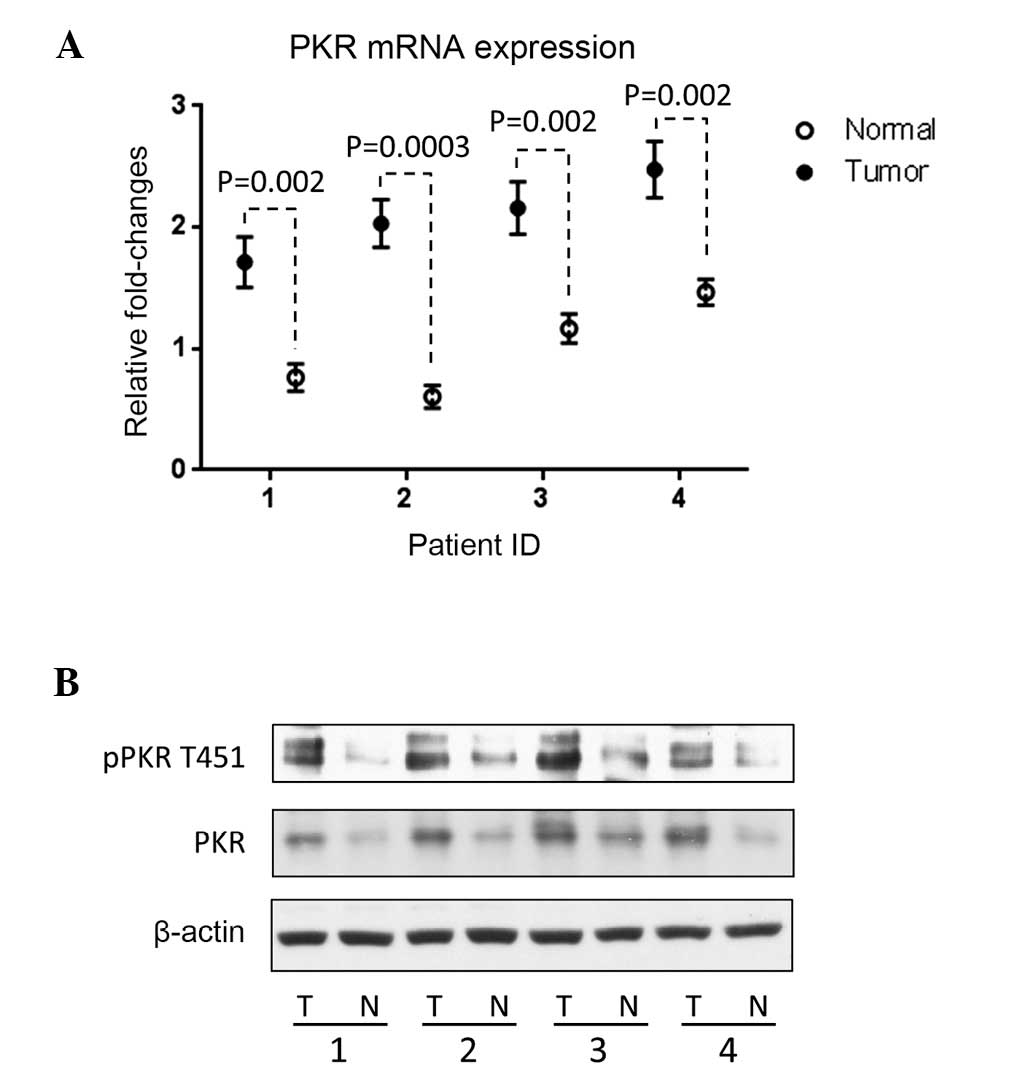
qPCR and western blotting were performed to measure
the PKR mRNA and protein expression levels respectively in tumor
tissues, using the adjacent normal tissue as a reference. PKR mRNA
(Fig. 1A) and protein expression
(Fig. 1B) was upregulated in all
four tumor samples, compared with the adjacent normal tissues. PKR
protein activity depends on phosphorylation at Thr451 (2,3);
therefore, the phosphorylation level of PKR at Thr451 was also
measured in all four tumor tissue samples. The results revealed
that the PKR protein activity, indicated by the phosphorylation
level at Thr451, was also higher in the liver tumor samples than
the normal tissues (Fig. 1B).
Statistically significant differences in PKR mRNA expression
between normal and tumor tissues were identified in all four
patients (P=0.002, P=0.0003, P=0.002 and P=0.002, respectively).
These results confirmed those of previous reports, which observed
elevated PKR expression levels in tumor tissues, including tissues
from liver tumors, and which revealed that the total
phosphorylation of PKR is also higher in tumor tissues (9–14). As
determined by these findings, the potential function of PKR in
regulating tumor cell phenotype, for instance, in modifying
proliferation and migration, was further analyzed.
PKR is involved in maintaining liver
cancer cell proliferation and migration
HepG2 cells served as a model for hepatocellular
carcinoma. Silencing PKR gene expression with PKR shRNA markedly
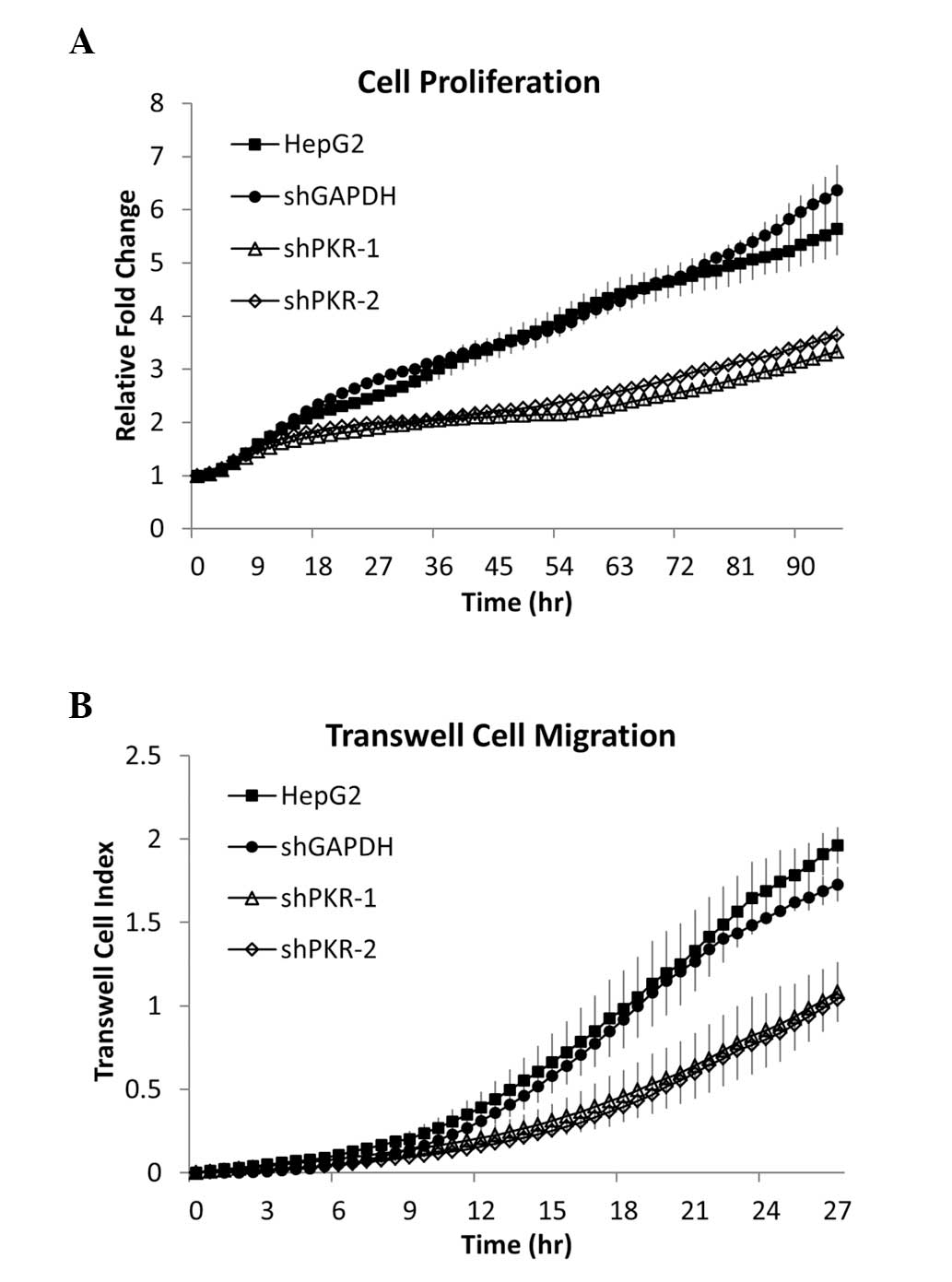
reduced the proliferation rate of HepG2 cells (Fig. 2A), suggesting that PKR is involved
in promoting HepG2 cell proliferation. In addition, as cell
migration is an early requirement for tumor metastasis and the rate
of migration indicates the aggressiveness of cancer cells, the
effect of PKR on cell migration was examined with in vitro
Transwell migration assays. Silencing PKR with shRNA markedly
suppressed HepG2 cell Transwell migration (Fig. 2B). To further investigate the role
of PKR in promoting HepG2 cell proliferation and Transwell
migration, PKR expression following gene silencing was rescued.
Proliferation and migration were completely restored by rescuing
PKR expression in HepG2 cells (Fig. 4B
and C). The results indicate that PKR is central in promoting
and maintaining HepG2 cell proliferation rates and migration
through micrometer pores. Thus, PKR may be involved in maintaining
liver cancer cell proliferation and migration, suggesting a
potential tumorigenic role for PKR in liver tumor cells.
PKR is involved in liver cancer
tumorigenesis
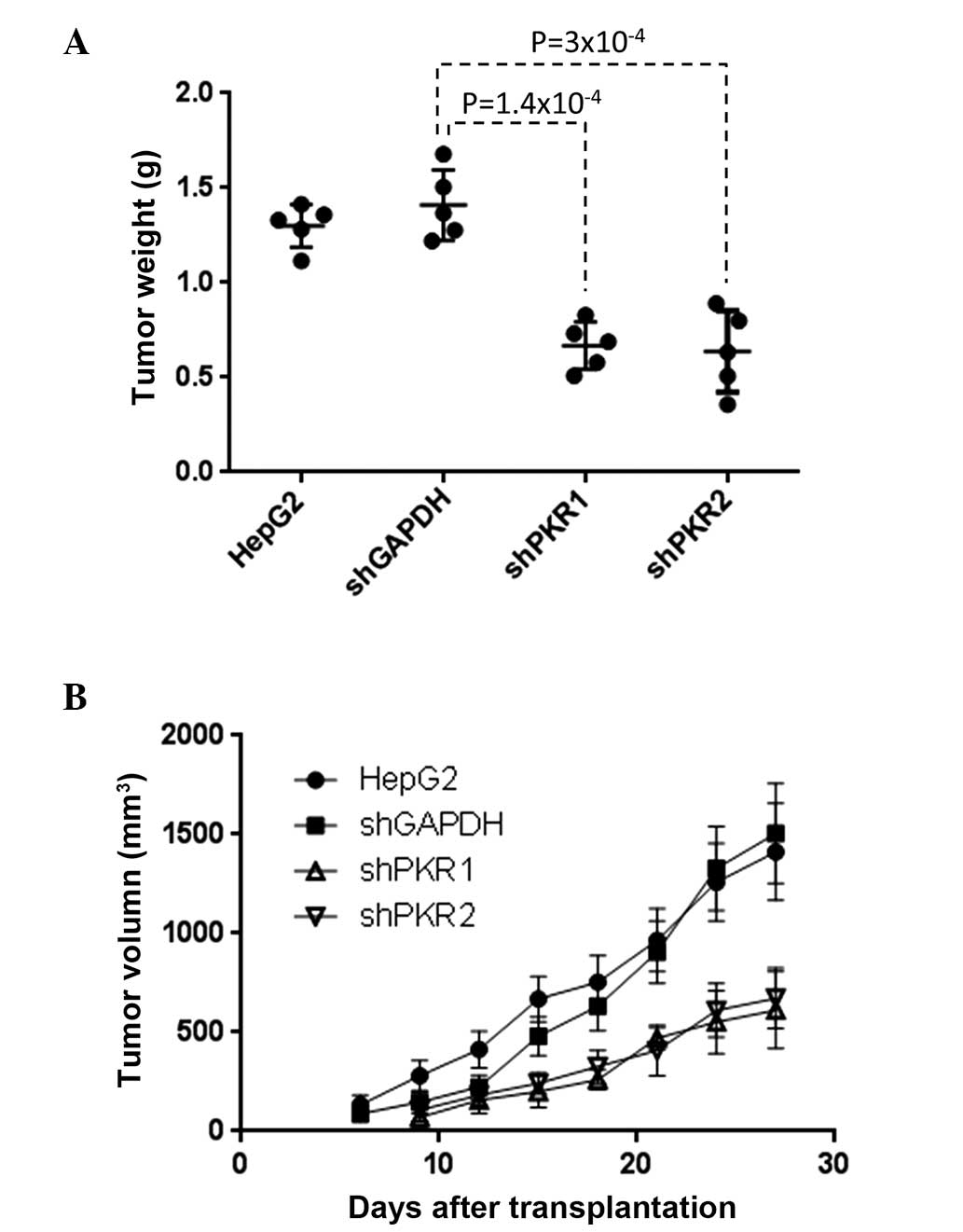
To examine the role of PKR in tumorigenesis in
vivo, xenograft transplantation experiments were performed in
mice. Vector-based PKR shRNA was used to prepare the HepG2 cell
line that stably expresses shPKR and therefore silences PKR
long-term. These cells were then subcutaneously injected into nude
mice (CD1−/−). Compared with the transplantation of
regular HepG2 cells and HepG2 cells that stably expressed empty
vector, transplantation of the cells that stably expressed shPKR
resulted in markedly slower tumor growth and smaller tumor size
four weeks after transplantation (Fig.
3). This clearly demonstrates that PKR exerts an important role
in promoting tumor development in vivo.
Overall, we have shown that PKR, which is
upregulated in primary liver tumors, is involved in maintaining
HepG2 cell proliferation and migration, and also exert a key role
in HepG2 cell tumorigenesis in vivo. However, the mechanisms
by which PKR regulates cell proliferation and migration, as shown
in Fig. 2, remains unclear.
PKR mediates HepG2 cell proliferation and
migration through STAT3
Previous studies have identified multiple downstream
targets of PKR, including eIF-2a, NF-κB and c-Jun N-terminal kinase
(3,15–17).
Silencing PKR has been shown to reduce Bcl-2 expression levels
through NF-κB. This was suggested to be the mechanism of
PKR-regulated cellular apoptosis. Indeed, the NF-κB signaling
pathway has been shown to be a key regulator of human
hepatocellular carcinoma development (24,25),
through various mechanisms. In addition to NF-κB, other
transcription factors, such as STAT3, have also been suggested to
promote the development of liver cancer (26). High STAT3 activity levels, which
depend on STAT3 phosphorylation, have been observed in primary
liver cancer (21). In addition,
hepatocyte-specific STAT3-deficient mice exhibited markedly greater
resistance to hepatocellular carcinoma and the tumor sizes were
evidently smaller, suggesting that STAT3 is crucial in promoting
hepatocellular carcinoma cell proliferation and/or survival
(22). Notably, PKR has been
demonstrated to directly bind with STAT3 and regulate STAT3
transcriptional activity, although whether PKR activates or
suppresses STAT3 remains controversial (19,27).
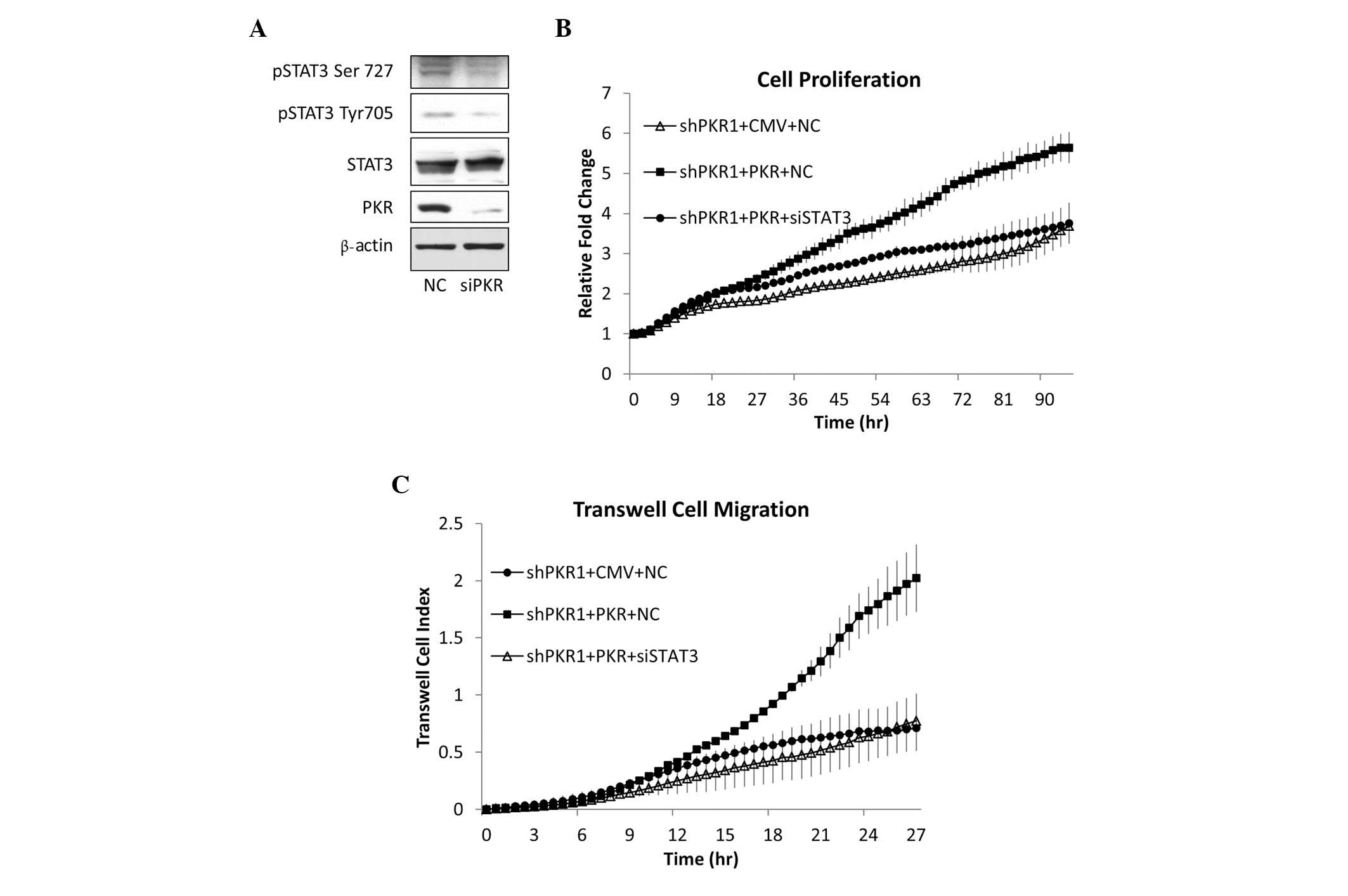
In the present study, the effect of PKR on
phosphorylation of STAT3 was analyzed. Silencing PKR gene
expression in HepG2 cells with siRNA reduced STAT3 phosphorylation
at Tyr705 and Ser727 (Fig. 4A).
Therefore, in HepG2 hepatocellular carcinoma cells, PKR positively
regulates STAT3 phosphorylation, a process hypothesized to
determine the activity of STAT3. Whether STAT3 is involved in
mediating the positive effect of PKR in promoting tumor cell growth
was then investigated. As shown in Fig.
2, shRNA lentivirus-mediated PKR gene silencing markedly
reduced HepG2 cell proliferation and migration. Subsequent rescue
of PKR expression following PKR knock-down restored cell
proliferation and migration (Fig. 4B
and C). Furthermore, in PKR-restored HepG2 cells, STAT3
expression was silenced with STAT3 siRNA. This completely reversed
the effects of rescuing PKR expression on cell growth and migration
rates (Fig. 4B and C). These
results demonstrate that PKR is essential in maintaining the high
growth and migration rates of HepG2 cells, and that this effect
depends on a well-known oncogenic transcription factor, STAT3.
Discussion
Previously, PKR has been recognized as a
first-response protein upon viral infection, due to its activation
by double-stranded RNA, which initiates innate immune responses by
arresting general protein synthesis and inducing apoptosis during
viral infection (28). Studies in
other systems have revealed further important roles of PKR in
mediating multiple signaling pathways, such as NF-κB,
mitogen-activate protein kinases (MAPKs) and protein phosphatase 2A
(PP2A) (29–32). Therefore, PKR was suggested to exert
a key role in other diseases systems, including those of cancer.
Due to its function in phosphorylating eIF-2α and thereby
inhibiting general protein synthesis (3,5), PKR
has been suggested to act as a tumor suppressor by suppressing cell
growth and inducing apoptosis (6,7).
However, studies have shown that PKR may exert an antiapoptotic
role in tumor cells (15,16). Notably, the protein expression
levels and activity of PKR have been found to be upregulated in
tumor cells; for example, in human breast cancer (12), melanoma (13) and hepatocellular carcinoma cells
(9–11). However, few studies investigating
the function of PKR in tumor cell proliferation and migration have
been published. PKR may suppress cell proliferation (6,33), but
the exact effect and the underlying mechanism remain unknown. More
recent results in HCV-related HCC revealed that PKR promotes tumor
cell proliferation through c-Fos and c-Jun signaling (34). In the present study, the function of
PKR in promoting the cell proliferation, migration and,
furthermore, tumorigenesis of hepatocellular carcinoma cells was
demonstrated. The results also revealed that PKR activates STAT3, a
transcription factor associated with primary liver tumors, which is
suggested to promote tumor cell proliferation (21).
As a Ser/Thr protein kinase, PKR is able to mediate
multiple important signaling pathways (4) in addition to eIF-2α, by interacting
with proteins, such as NF-κB, MAPKs and PP2A (29–32).
Two independent studies have reported opposite effects of PKR on
STAT3 activity (19,20). In mouse embryonic fibroblasts, PKR
was shown to dephosphorylate STAT3 at Tyr705 by activating T-cell
protein-tyrosine phosphatase (20),
while in another study, STAT3 phosphorylation at Tyr705 and Ser727
were observed to be dependent on PKR and the corresponding
downstream target ERK (19). This
controversy has not been fully resolved, although the effect on
phosphorylation may be associated with the basal levels of PKR and
STAT3 (20). In the present study,
relatively middle-to-high PKR and STAT3 activity levels were
observed in HepG2 cells (Fig. 4A),
and silencing PKR resulted in reduction of STAT3 activity.
Previous results have also suggested an
antiapoptotic role of PKR in HepG2 cells (16). In the present study, by focusing on
a small area in which none of the cells undergo apoptosis during
the observation time, the non-apoptotic cells exhibited slower
proliferation. In addition, the overexpression of PKR by
transfecting the pCMV-PKR plasmid into HepG2 cells increased the
rate of proliferation, during which no apoptosis was observed in
either control or PKR-overexpressing cells. PKR as a protein
kinase, activates several transcription factors, including IRF-1,
p53 and NF-κB (17,18). The effect of PKR on apoptosis of
hepatocellular carcinoma cells depends on the transcription factor
NF-κB (16). Notably, multiple
transcriptional events have been identified in liver cancer,
including NF-κB and STAT3. Previous studies observed that STAT3 and
NF-κB activation are mutually exclusive in liver cancer tissues,
and the molecules are engaged in positive and negative crosstalk
(22,26). Considering the results of previous
studies, together with those of the current study, PKR may
positively regulate the two transcription factors in the same cell
context. Since the factors have different and potentially
complementary effects on tumor cell activities, including
apoptosis, proliferation and migration, further investigation into
whether and how PKR is involved in the crosstalk between NF-κB and
STAT3 may be required.
Acknowledgements
The present study was financially supported by the
General Hospital of Chinese PLA, the Tsinghua-Peking Center for
Life Sciences and the 1000 Talent Program (Youth Category).
References
|
1
|
Barber GN: The dsRNA-dependent protein
kinase, PKR and cell death. Cell Death Differ. 12:563–570.
2005.
|
|
2
|
Zhang F, Romano PR, Nagamura-Inoue T, et
al: Binding of double-stranded RNA to protein kinase PKR is
required for dimerization and promotes critical autophosphorylation
events in the activation loop. J Biol Chem. 276:24946–24958.
2001.
|
|
3
|
Taylor SS, Haste NM and Ghosh G: PKR and
eIF2alpha: integration of kinase dimerization, activation, and
substrate docking. Cell. 122:823–825. 2005.
|
|
4
|
Williams BR: Signal integration via PKR.
Sci STKE. 2001:re22001.
|
|
5
|
Stark GR, Kerr IM, Williams BR, Silverman
RH and Schreiber RD: How cells respond to interferons. Annu Rev
Biochem. 67:227–264. 1998.
|
|
6
|
Koromilas AE, Roy S, Barber GN, Katze MG
and Sonenberg N: Malignant transformation by a mutant of the
IFN-inducible dsRNA-dependent protein kinase. Science.
257:1685–1689. 1992.
|
|
7
|
Meurs EF, Galabru J, Barber GN, Katze MG
and Hovanessian AG: Tumor suppressor function of the
interferon-induced double-stranded RNA-activated protein kinase.
Proc Natl Acad Sci USA. 90:232–236. 1993.
|
|
8
|
Roh V, Laemmle A, Von Holzen U, et al:
Dual induction of PKR with E2F-1 and IFN-alpha to enhance gene
therapy against hepatocellular carcinoma. Cancer Gene Ther.
15:636–644. 2008.
|
|
9
|
Hiasa Y, Kamegaya Y, Nuriya H, et al:
Protein kinase R is increased and is functional in hepatitis C
virus-related hepatocellular carcinoma. Am J Gastroenterol.
98:2528–2534. 2003.
|
|
10
|
Delhem N, Sabile A, Gajardo R, et al:
Activation of the interferon-inducible protein kinase PKR by
hepatocellular carcinoma derived-hepatitis C virus core protein.
Oncogene. 20:5836–5845. 2001.
|
|
11
|
Mohamed AA, Nada OH and El Desouky MA:
Implication of protein kinase R gene quantification in hepatitis C
virus genotype 4 induced hepatocarcinogenesis. Diagn Pathol.
7:1032012.
|
|
12
|
Kim SH, Forman AP, Mathews MB and Gunnery
S: Human breast cancer cells contain elevated levels and activity
of the protein kinase, PKR. Oncogene. 19:3086–3094. 2000.
|
|
13
|
Kim SH, Gunnery S, Choe JK and Mathews MB:
Neoplastic progression in melanoma and colon cancer is associated
with increased expression and activity of the interferon-inducible
protein kinase, PKR. Oncogene. 21:8741–8748. 2002.
|
|
14
|
Delgado André N and De Lucca FL: Knockdown
of PKR expression by RNAi reduces pulmonary metastatic potential of
B16-F10 melanoma cells in mice: Possible role of NF-kappaB. Cancer
Lett. 258:118–125. 2007.
|
|
15
|
Donzé O, Deng J, Curran J, et al: The
protein kinase PKR: a molecular clock that sequentially activates
survival and death programs. EMBO J. 23:564–571. 2004.
|
|
16
|
Yang X and Chan C: Repression of PKR
mediates palmitate-induced apoptosis in HepG2 cells through
regulation of Bcl-2. Cell Res. 19:469–486. 2009.
|
|
17
|
Kumar A, Yang YL, Flati V, et al:
Deficient cytokine signaling in mouse embryo fibroblasts with a
targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB.
EMBO J. 16:406–416. 1997.
|
|
18
|
Cuddihy AR, Li S, Tam NW, et al:
Double-stranded-RNA-activated protein kinase PKR enhances
transcriptional activation by tumor suppressor p53. Mol Cell Biol.
19:2475–2484. 1999.
|
|
19
|
Deb A, Zamanian-Daryoush M, Xu Z, Kadereit
S and Williams BR: Protein kinase PKR is required for
platelet-derived growth factor signaling of c-fos gene expression
via Erks and Stat3. EMBO J. 20:2487–2496. 2001.
|
|
20
|
Wang S, Raven JF, Baltzis D, et al: The
catalytic activity of the eukaryotic initiation factor-2alpha
kinase PKR is required to negatively regulate Stat1 and Stat3 via
activation of the T-cell protein-tyrosine phosphatase. J Biol Chem.
281:9439–9449. 2006.
|
|
21
|
Calvisi DF, Ladu S, Gorden A, et al:
Ubiquitous activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006.
|
|
22
|
He G, Yu GY, Temkin V, et al: Hepatocyte
IKKbeta/NF-kappaB inhibits tumor promotion and progression by
preventing oxidative stress-driven STAT3 activation. Cancer Cell.
17:286–297. 2010.
|
|
23
|
Carro MS, Lim WK, Alvarez MJ, et al: The
transcriptional network for mesenchymal transformation of brain
tumours. Nature. 463:318–325. 2010.
|
|
24
|
Maeda S, Kamata H, Luo JL, Leffert H and
Karin M: IKKbeta couples hepatocyte death to cytokine-driven
compensatory proliferation that promotes chemical
hepatocarcinogenesis. Cell. 121:977–990. 2005.
|
|
25
|
Maeda S, Hikiba Y, Sakamoto K, et al:
Ikappa B kinasebeta/nuclear factor-kappaB activation controls the
development of liver metastasis by way of interleukin-6 expression.
Hepatology. 50:1851–1860. 2009.
|
|
26
|
He G and Karin M: NF-kappaB and STAT3 -
key players in liver inflammation and cancer. Cell Res. 21:159–168.
2011.
|
|
27
|
Papadakis AI, Paraskeva E, Peidis P, et
al: eIF2α Kinase PKR Modulates the Hypoxic Response by
Stat3-Dependent Transcriptional Suppression of HIF-1α. Cancer Res.
70:7820–7829. 2010.
|
|
28
|
Proud CG: PKR: a new name and new roles.
Trends Biochem Sci. 20:241–246. 1995.
|
|
29
|
Kumar A, Haque J, Lacoste J, Hiscott J and
Williams BR: Double-stranded RNA-dependent protein kinase activates
transcription factor NF-kappa B by phosphorylating I kappa B. Proc
Natl Acad Sci USA. 91:6288–6292. 1994.
|
|
30
|
Gil J, Alcamí J and Esteban M: Activation
of NF-kappa B by the dsRNA-dependent protein kinase, PKR involves
the I kappa B kinase complex. Oncogene. 19:1369–1378. 2000.
|
|
31
|
Zhou HR, Lau AS and Pestka JJ: Role of
double-stranded RNA-activated protein kinase R (PKR) in
deoxynivalenol-induced ribotoxic stress response. Toxicol Sci.
74:335–344. 2003.
|
|
32
|
Xu Z and Williams BR: The B56alpha
regulatory subunit of protein phosphatase 2A is a target for
regulation by double-stranded RNA-dependent protein kinase PKR. Mol
Cell Biol. 20:5285–5299. 2000.
|
|
33
|
Donzé O, Jagus R, Koromilas AE, Hershey JW
and Sonenberg N: Abrogation of translation initiation factor eIF-2
phosphorylation causes malignant transformation of NIH 3T3 cells.
EMBO J. 14:3828–3834. 1995.
|
|
34
|
Watanabe T, Hiasa Y, Tokumoto Y, et al:
Protein kinase R modulates c-Fos and c-Jun signaling to promote
proliferation of hepatocellular carcinoma with hepatitis C virus
infection. PLoS One. 8:e677502013.
|