Introduction
Trauma may produce direct organ system injury or be
accompanied by hemodynamic alterations, which may result in organ
dysfunction/failure. Traumatic injury may also initiate an acute
inflammatory response in injured tissues or organs, which may
induce an uncontrolled systemic inflammatory response and result in
multiple organ dysfunction syndrome (MODS), previously known as
multiple organ failure or multisystem organ failure. In general,
the acute or systematic inflammatory response is considered to be
mediated through the interactions among cytokines, including the
activation of tumor necrosis factor (TNF) and the corresponding
receptors and neuroendocrine pathways (1–3).
TNF-α is a key cytokine involved in the generation
of the acute inflammatory response (1,3). This
inflammatory cytokine is primarily produced by immune cells, such
as monocytes and macrophages, but a number of non-immune cell
types, including fibroblasts, neurons, keratinocytes and smooth
muscle cells, also produce TNF. TNF-α acts as a key intermediary in
the local inflammatory immune response and is an acute-phase
protein that initiates a cascade of cytokines. Furthermore, high
levels of TNF result in increased vascular permeability, thereby
recruiting macrophages and neutrophils to the site of injury and/or
infection (2,3). The action of TNF-α is mediated via
cell-surface TNF receptors (TNFRs) (4). Two distinct members of the TNFR family
are currently recognized: TNFR1, also known as p55, and TNFR2,
termed p75 (5–7). TNFR1 is expressed constitutively in
the majority of cell types, whereas TNFR2 expression is restricted
to hematopoietic cells and discriminates between the murine and
human forms of TNF-α (8). TNFR2
expression has been shown to be induced by TNF-α, IL-1 and
interferon-γ in rat primary astrocytes (9). The two receptors may function
individually or synergistically to mediate the biological activity
of TNF-α.
The plasma levels of TNF and the respective
receptors are increased in response to severe trauma (10–13).
By contrast, results for elevated TNF-α levels following trauma are
commonly negative; low TNF-α levels have been reported to promote
the remodeling or replacement of injured tissue by stimulating
fibroblast growth (14–20). Additional beneficial functions of
TNF-α include its involvement in the immune response to bacterial
and certain fungal, viral and parasitic invasions, as well as in
the necrosis of specific tumors (21). The present study aimed to
investigate the plasma levels of TNF-α and the corresponding
receptors, as well as the expression levels of TNFRs on leukocytes
in the early phase following multiple traumatic injuries and up to
five days intensive care. The objective was to analyze the
time-dependent correlations between these immunological parameters
and injury severity.
Subjects and methods
Patients
A total of 60 trauma patients (42 males and 18
females, aged 18 to 72 years) were included in the study. These
patients were treated at the Department of Emergency Surgery,
Jinshan Hospital, Fudan University (Shanghai, China) over a time
period of three years (2008–2011). On hospital admission, the
injury severity score (ISS) was recorded for the assessment of
injury severity (11,18). Patients who suffered from chronic
immune deficiency or who were pregnant were excluded from the
study. As determined by the mean ISS, the patients were divided
into the following three groups (n=20 patients per group): Low ISS
(L-ISS; 9≤ ISS <16), medium ISS (M-ISS; 16≤ ISS <25) and high
ISS (H-ISS; ISS ≥25). The control group included 20 healthy
volunteers between the ages of 18 and 25 years. This study was
conducted in accordance with the Declaration of Helsinki and with
approval from the Ethics Committee of Jinshan Hospital, Fudan
University. Written informed consent was obtained from all
participants.
Blood samples
Venous blood was collected from the patients and
healthy volunteers, and was maintained in tubes with
ethylenediaminetetraacetate anticoagulant. A volume of 10 ml blood
was sampled from all patients at five defined time points. The
first sample was obtained upon hospital admission by the emergency
physician (usually within 2 h of injury) and the second sample was
obtained 6–8 h after admission (or 8–10 h after trauma). Further
blood samples were collected 24 h after admission, and then on days
3 and 5 post-admission. Following cooling centrifugation (20 min at
4°C and 1,500 × g), the plasma was aliquoted and frozen at −70°C in
250-μl aliquots for TNF-α and soluble TNFR1/TNFR2 (sTNFR1/sTNFR2)
expression analysis.
Measurement of TNF-α, sTNFR1 and sTNFR2
levels
The concentrations of TNF-α and the corresponding
receptors in the plasma were determined by enzyme-linked
immunosorbent assay (ELISA) techniques. The analyses were performed
in duplicate using standardized, commercially available enzyme
immunoassay kits according to the manufacturers’ instructions. A
human TNF-α chemiluminescent ELISA kit (Thermo Fisher Scientific,
Waltham, MA, USA) and sTNFR ELISA kits (Abcam, Cambridge, MA, USA)
were employed. The sensitivities of the assays were <1 pg/ml for
TNF-α and sTNFR1, and <5 pg/ml for sTNFR2. The control levels of
the cytokine and the receptors were determined from the healthy
volunteers.
Flow cytometric detection of TNFRs in
leukocytes
Whole blood samples were collected by standard
syringe venipuncture upon hospital admission (within 2 h of injury)
and mixed with anticoagulant (heparin, 10 IU/ml). Control samples
were collected from matched healthy volunteers at similar time
points in the day to when patient samples were collected. The cell
samples were stained in the dark on ice for 60 min with
affinity-purified rabbit polyclonal antibody for TNFR1 (1:500;
Abcam), mouse monoclonal antibody for TNFR2 (MR2-1; 1:1,000; Abcam)
or the appropriate controls (isotype or unstained).
Antibody-binding was visualized with anti-rabbit or -mouse IgG
conjugated to a fluorophore (60 min on ice) (Thermo Fisher
Scientific). The samples were analyzed using a Gallios flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA) and CXP Analysis
software (v2.1; Applied Cytometry Systems, Dinnington, UK). The
expression levels of TNFRs in neutrophils, lymphocytes and
monocytes were assessed, and are expressed as the mean fluorescence
intensity following subtraction of the isotype control value.
Statistical analysis
Differences among groups were compared using one-way
or two-way analysis of variance (for degree of injury and leucocyte
type) with Bonferroni’s post hoc analysis. The time-course changes
in the expression levels of TNF-α and the corresponding receptors
were analyzed by linear regression. The correlation coefficients
(r) were calculated using Spearman’s rank test. P<0.05 was
considered to indicate a statistically significant difference and
data are expressed as the mean ± standard deviation. SPSS 11.0
(SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 5 (GraphPad, La
Jolla, CA, USA) were used for data analysis.
Results
Plasma TNF-α levels in trauma
patients
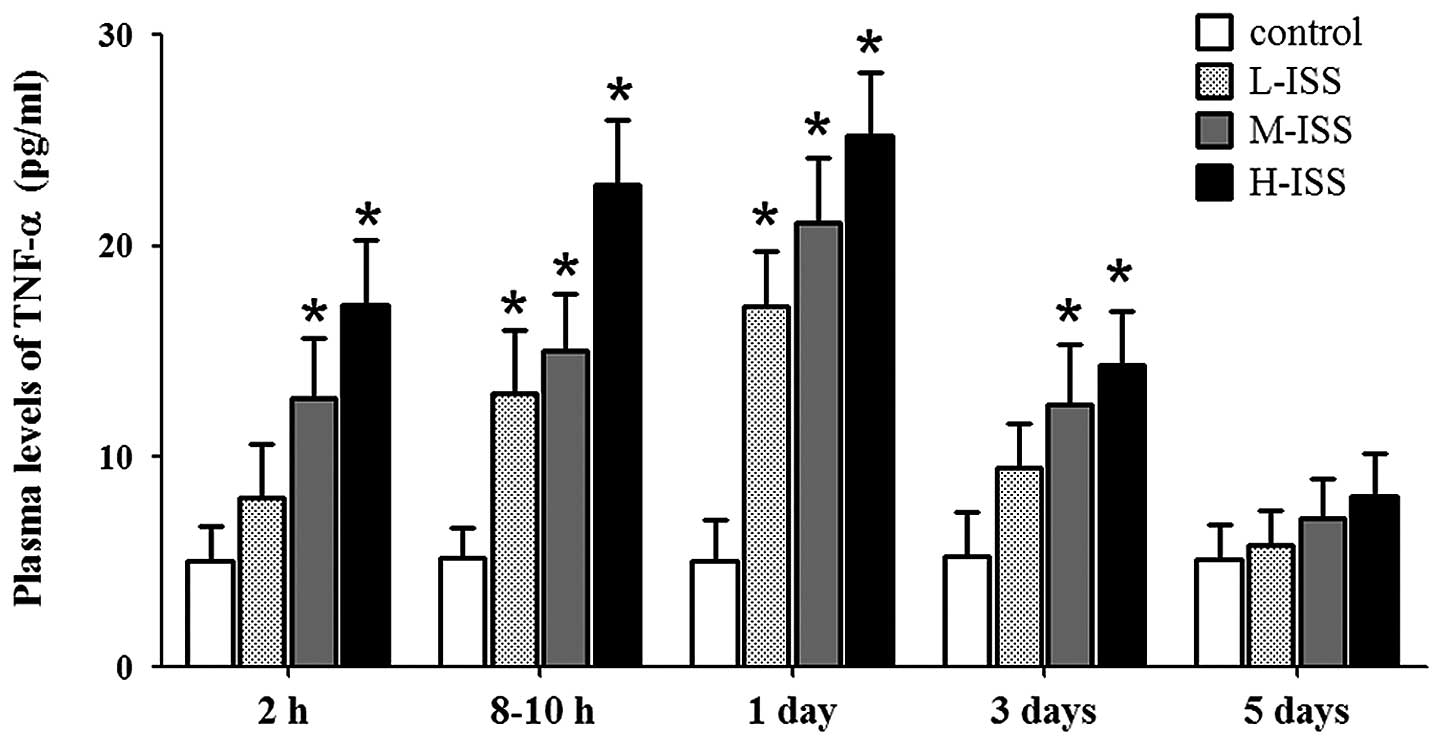
Plasma TNF-α levels in all three trauma groups
(L-ISS, M-ISS and H-ISS) were significantly elevated following
injury (usually within 2 h trauma) as compared with the healthy
controls (5.05 pg/ml in controls versus 8.07–17.23 pg/ml in
patients; P<0.05; Fig. 1). Peak
plasma TNF-α levels were detected 24 h after injury and the levels
remained significantly elevated until up to the third day following
trauma. At five days after injury, plasma TNF-α levels had
gradually returned to the normal levels (Fig. 1). Furthermore, the TNF-α levels were
significantly correlated with the severity of injury, as indicated
by the ISS (r=0.78, P<0.0001).
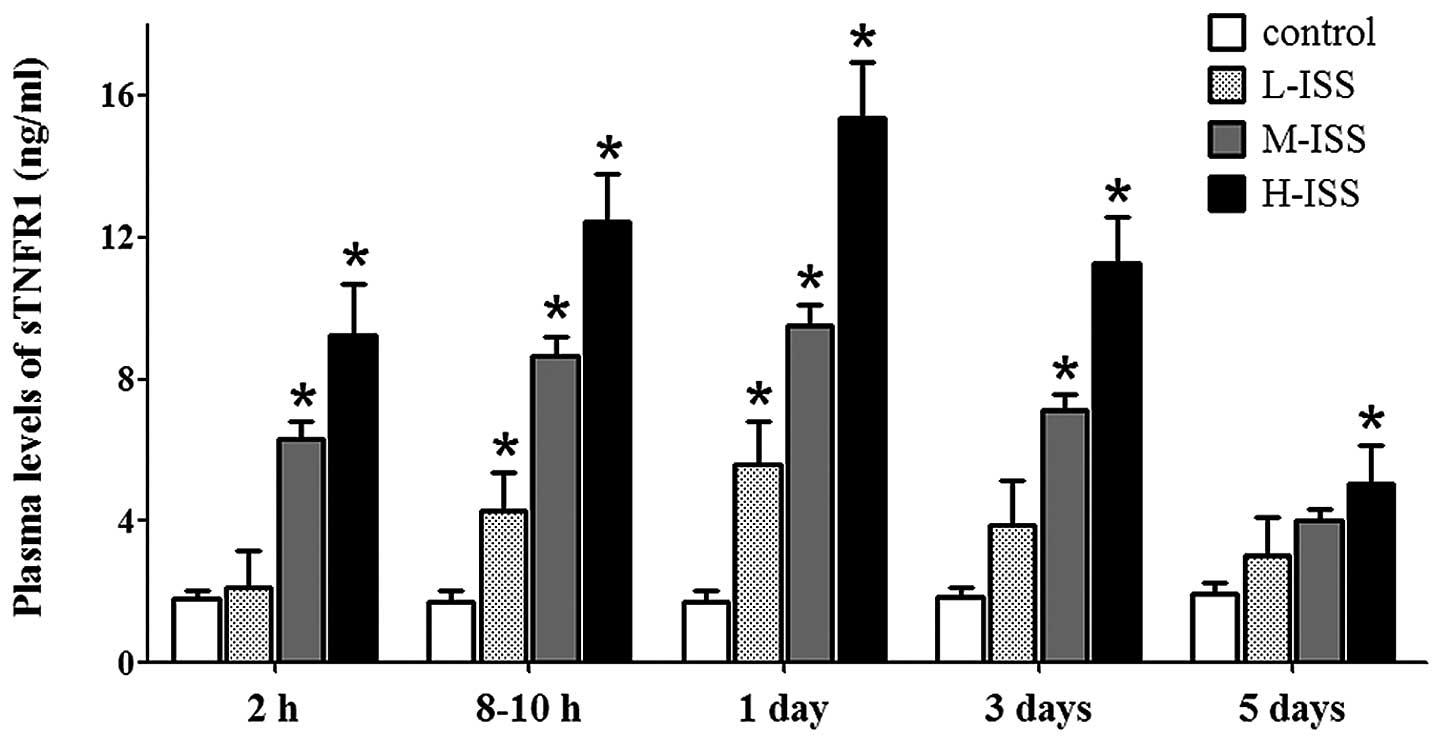
sTNFR plasma levels in patients with
severe trauma
Within 2 h of injury, the sTNFR1 and sTNFR2 plasma
levels were significantly elevated in all trauma groups compared
with the normal controls (1.83±0.23 and 1.46±0.42 pg/ml for sTNFR1
and sTNFR2 in the controls, respectively). The patient group with
the highest severity score (H-ISS group) exhibited the highest
sTNFR1 and sTNFR2 plasma levels in the early phase following
trauma. Elevated expression levels of soluble receptors were also
observed 8–10 h after injury and 1, 3 and 5 days after trauma.
sTNFR1 expression reached peak levels one day after trauma, which
was gradually reduced and returned to normal five days after trauma
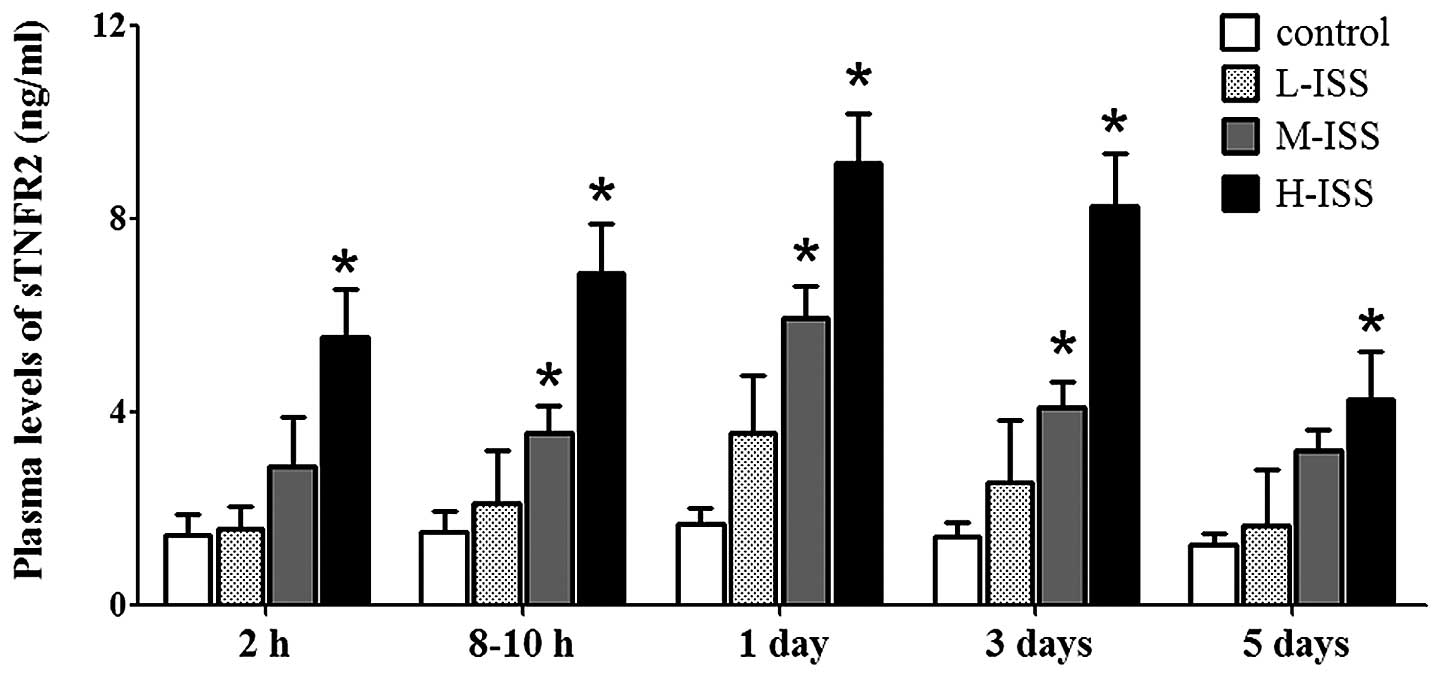
(Fig. 2). Although increased levels
of sTNFR2 were also detected in trauma patients compared with the
controls, sTNFR2 levels were elevated for variable periods of time
and were dependent on the severity of injury (Fig. 3). The statistical analysis suggested
a significant correlation between the plasma levels of the sTNFRs
and the severity of traumatic injury upon hospital admission
(sTNFR1: r=0.89, P<0.0001 and sTNFR2: r=0.92, P<0.0001) as
well as at the other four time points (data not shown).
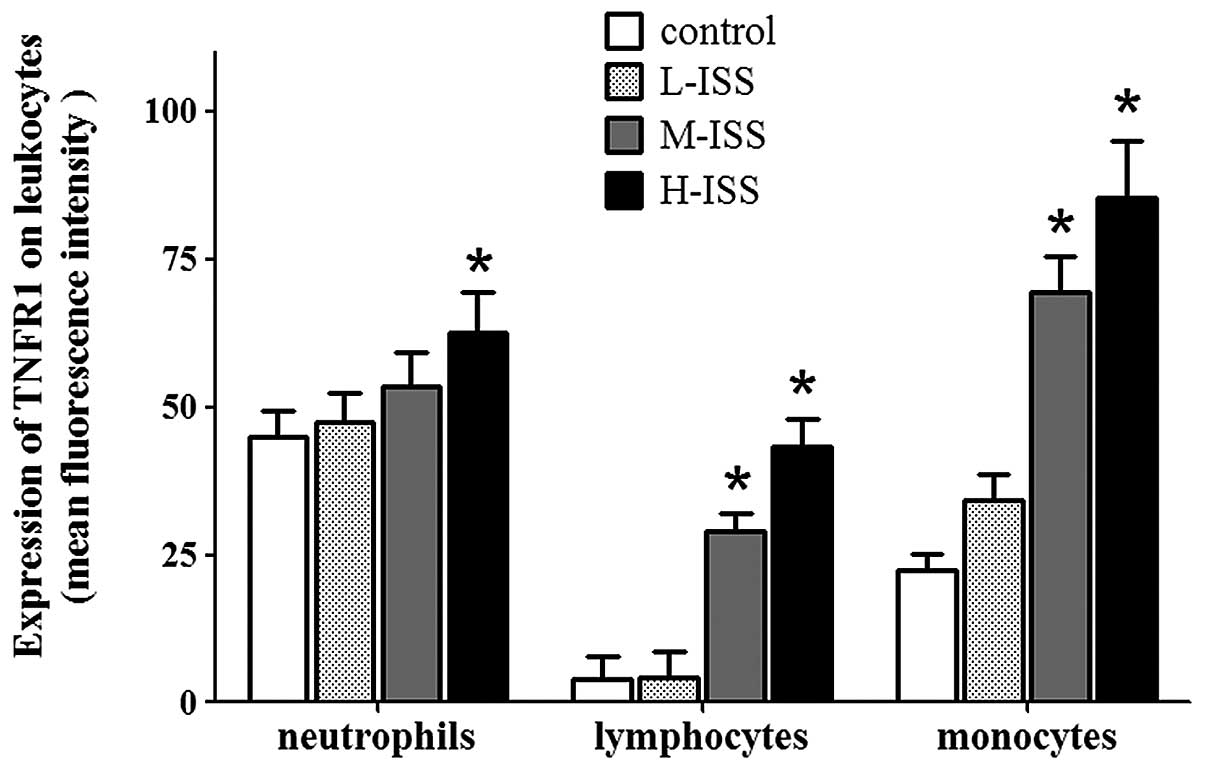
Expression levels of TNFRs on
leukocytes
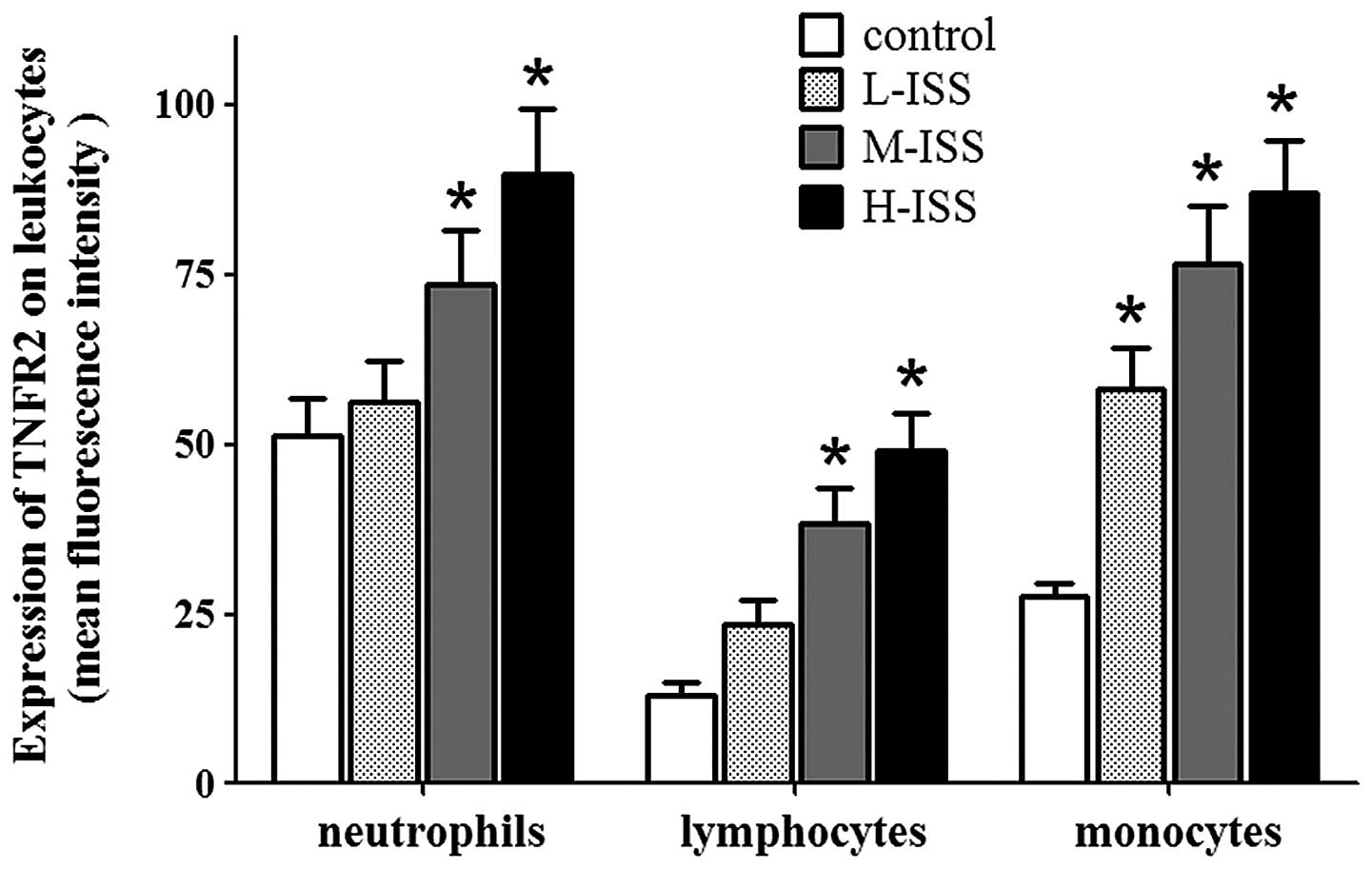
TNFR1 and TNFR2 were detected on leukocytes under
control conditions, with high expression levels in neutrophils,
moderate expression levels in monocytes and lower expression levels
in lymphocytes. Traumatic injury resulted in enhanced TNFR1 and
TNFR2 expression levels, with the highest expression levels in the
H-ISS patient group. In particular, the injury-induced increase in
expression levels of TNFRs was marked in monocytes and lymphocytes
in the early phases following trauma. Furthermore, the increases in
TNFR1 expression levels in monocytes and lymphocytes were
significantly correlated with the severity of traumatic injury
(monocytes: r=0.89, P<0.0001 and lymphocytes: r=0.93,
P<0.0001; Fig. 4). A significant
correlation between TNFR2 expression levels and injury severity was
also detected in monocytes (r=0.89, P<0.0001) and lymphocytes
(r=0.91, P<0.0001) (Fig. 5).
Discussion
In the present study, the time-course changes in the
expression levels of plasma TNF-α and the corresponding TNFR1 (p55)
and TNFR2 (p75) receptors were examined in 60 trauma patients.
Traumatic injury was found to elevate cytokine and soluble receptor
plasma levels in the early phases of trauma, which remained
elevated for up to the third to fifth day after the trauma. The
TNFR expression levels on the surfaces of freshly harvested
leukocytes in response to traumatic injury were also quantitatively
analyzed; increased expression levels of TNFRs were found to be
particularly marked in monocytes and lymphocytes. Notably, the
plasma levels of the cytokine and the respective receptors, as well
as the surface expression levels of the receptors on the
leukocytes, were correlated with injury severity, as indicated by
the ISS.
TNF-α is particularly important as part of the
inflammatory response. This cytokine initiates the activation of
several other cytokines and growth factors, as well as the
recruitment of certain immune cells. TNF-α has been reported to be
released more rapidly than other proinflammatory cytokines
(22). In general, elevated TNF-α
levels subsequent to trauma are harmful to the body (14–20),
and increased TNF-α levels have been shown to induce central
sensitization and hyperalgesia by increasing excitatory synaptic
transmission (23). However,
following traumatic injury to the brain, TNF-α exerts a
neuroprotective role by contributing to neuroanatomical plasticity
(24). In the present study, plasma
TNF-α levels were detected to be elevated in all trauma patients
immediately following trauma in comparison with healthy controls.
Specifically, significant injury-induced elevation of TNF-α levels
was detected up to three days subsequent to trauma. These results
are consistent with those of Spielmann et al (20), in which a continuous and significant
increase in TNF levels was identified in patients 4 h after trauma.
In certain early studies, TNF-α was reported to be either
unmeasurable (10,16,18) or
not correlated with the severity of injury, and TNF-α is known as a
sensitive parameter during sample processing. In the present study,
plasma samples were collected immediately following blood
withdrawal by cooling, centrifugation and freezing at −70°C. A
highly sensitive ELISA kit was employed using enhanced
chemiluminescence to detect TNF levels. Peak plasma TNF-α levels
were detected 24 h after injury. Furthermore, the TNF-α levels
remained significantly elevated for up to three days following
trauma. At five days after injury, the plasma TNF-α levels
gradually returned to normal. The data further suggest that the
trauma-induced elevation in TNF-α levels was associated with the
ISS.
Administration of TNF-α has observed to increase the
expression levels of sTNFR1 and sTNFR2 in experimental conditions
(25,26), suggesting a TNF-α-induced
inflammatory response. TNF-α receptors may also be induced by
direct damage to tissue, disturbed macro- and microcirculation with
subsequent ischemia and/or reperfusion, and psychoneurological
stimuli (27). In the present
study, TNFRs were detected to be elevated on leukocytes as well as
in the plasma of trauma patients, compared with healthy controls.
Leukocytes are key anti-infectious agents in host defense, and
neutrophils and monocytes are important inflammatory cells that
mediate tissue damage. A flow cytometric method was established
that enabled the quantitative evaluation of receptor expression
levels on leukocytes in a single-cell suspension. The p55 and p75
receptors were found to be highly expressed on leukocytes,
particularly on monocytes and lymphocytes, in response to traumatic
injury. Notably, the increases in injury-induced TNFR expression
levels were positively correlated with the severity of trauma. In
the plasma, increased levels of TNFRs were also detected in the
very early injury phase, and were associated with the severity of
trauma. Furthermore, the expression levels of the two soluble
receptors were correlated with the ISS in the early phase following
trauma. These results confirm those of other studies, which
demonstrated that the two receptor subtypes were involved in the
inflammatory response. In early injury, sTNFR1 levels have been
reported to be elevated directly following the accident, whereas
sTNFR2 levels were increased after 24 h (20). The data from the present study
indicated elevated sTNFR1 as well as sTNFR2 levels on admission to
hospital, and receptor levels correlated significantly with the
ISS. These data are consistent with those from other studies
(28,29), in which soluble TNF-α receptors were
shown to be more elevated in patients with lower survival rates. In
addition, Froon et al (30)
reported a significant elevation in sTNFR1 receptor expression
levels during sepsis.
MODS is generally recognized as a predominant cause
of mortality in trauma, systemic inflammatory response syndrome
(SIRS) and a number of other critical illnesses. The systemic
inflammatory response is rapidly followed in the majority of
patients by a compensatory anti-inflammatory response, signifying
an attempt to limit the SIRS response. Organ dysfunction is likely
to ensue during an excessive inflammatory reaction. The patient is
also at risk of opportunistic or secondary infection during an
excessive anti-inflammatory response. Numerous potential humoral,
cellular and exogenous mediators are involved in the pathogenesis
of MODS/multiple organ failure, and various pathways that result in
organ system dysfunction/damage (31). Among the potential
mediators/pathways, TNF and the respective receptor signaling
pathway may be critical in the pathogenesis of MODS. The data from
the present study suggest that TNFRs are positively associated with
the severity of injury. Therefore, inhibiting the activity of TNF-α
and other cytokines as a therapy for trauma is of interest. Trials
that aim to block, neutralize or remove the potential inflammatory
mediators have shown success in several preclinical models of
trauma, including spinal cord injury (32), brain injury (33) and liver injury (34), as well as in patients with chronic
neurological dysfunction following stroke or traumatic brain injury
(35).
In conclusion, the data from the present study
demonstrate increased plasma levels of TNF-α and sTNFRs in response
to severe traumatic injury. The study also provides invaluable data
regarding TNFR expression in leukocytes, by quantitative assessment
of the receptors on freshly harvested neutrophils, lymphocytes and
monocytes using flow cytometry. The results highlight the potential
correlation between TNFR expression levels and injury severity,
supporting the hypothesis that an auto-destructive inflammatory
response may cause trauma-initiated organ failure.
Acknowledgements
This study was supported by the Shanghai Health
Bureau (grant no. 20114261).
References
|
1
|
Lenz A, Franklin GA and Cheadle WG:
Systemic inflammation after trauma. Injury. 38:1336–1345. 2007.
|
|
2
|
Esposito E and Cuzzocrea S: TNF-alpha as a
therapeutic target in inflammatory diseases, ischemia-reperfusion
injury and trauma. Curr Med Chem. 16:3152–3167. 2009.
|
|
3
|
Woodcock T and Morganti-Kossmann MC: The
role of markers of inflammation in traumatic brain injury. Front
Neurol. 4:182013.
|
|
4
|
Vandenabeele P, Declercq W, Beyaert R and
Fiers W: Two tumour necrosis factor receptors: structure and
function. Trends Cell Biol. 5:392–399. 1995.
|
|
5
|
Loetscher H, Pan YC, Lahm HW, Gentz R,
Brockhaus M, Tabuchi H and Lesslauer W: Molecular cloning and
expression of the human 55 kd tumor necrosis factor receptor. Cell.
61:351–359. 1990.
|
|
6
|
Smith CA, Davis T, Anderson D, et al: A
receptor for tumor necrosis factor defines an unusual family of
cellular and viral proteins. Science. 248:1019–1023. 1990.
|
|
7
|
Goodwin RG, Anderson D, Jerzy R, et al:
Molecular cloning and expression of the type 1 and type 2 murine
receptors for tumor necrosis factor. Mol Cell Biol. 11:3020–3026.
1991.
|
|
8
|
Tartaglia LA, Weber RF, Figari IS,
Reynolds C, Palladino MA Jr and Goeddel DV: The two different
receptors for tumor necrosis factor mediate distinct cellular
responses. Proc Natl Acad Sci USA. 88:9292–9296. 1991.
|
|
9
|
Choi SJ, Lee KH, Park HS, Kim SK, Koh CM
and Park JY: Differential expression, shedding, cytokine regulation
and function of TNFR1 and TNFR2 in human fetal astrocytes. Yonsei
Med J. 46:818–826. 2005.
|
|
10
|
Cinat M, Waxman K, Vaziri ND, Daughters K,
Yousefi S, Scannell G and Tominaga GT: Soluble cytokine receptors
and receptor antagonists are sequentially released after trauma. J
Trauma. 39:112–120. 1995.
|
|
11
|
Ertel W, Keel M, Bonaccio M, Steckholzer
U, Gallati H, Kenney JS and Trentz O: Release of anti-inflammatory
mediators after mechanical trauma correlates with severity of
injury and clinical outcome. J Trauma. 39:879–887. 1995.
|
|
12
|
Stein DM, Lindell A, Murdock KR, et al:
Relationship of serum and cerebrospinal fluid biomarkers with
intracranial hypertension and cerebral hypoperfusion after severe
traumatic brain injury. J Trauma. 70:1096–1103. 2011.
|
|
13
|
Dalgard CL, Cole JT, Kean WS, et al: The
cytokine temporal profile in rat cortex after controlled cortical
impact. Front Mol Neurosci. 5:62012.
|
|
14
|
Hoch RC, Rodriguez R, Manning T, Bishop M,
Mead P, Shoemaker WC and Abraham E: Effects of accidental trauma on
cytokine and endotoxin production. Crit Care Med. 21:839–845.
1993.
|
|
15
|
Rabinovici R, John R, Esser KM, Vernick J
and Feuerstein G: Serum tumor necrosis factor-alpha profile in
trauma patients. J Trauma. 35:698–702. 1993.
|
|
16
|
Tan LR, Waxman K, Scannell G, Ioli G and
Granger GA: Trauma causes early release of soluble receptors for
tumor necrosis factor. J Trauma. 34:634–638. 1993.
|
|
17
|
Cinat ME, Waxman K, Granger GA, Pearce W,
Annas C and Daughters K: Trauma causes sustained elevation of
soluble tumor necrosis factor receptors. J Am Coll Surg.
179:529–537. 1994.
|
|
18
|
Svoboda P, Kantorová I and Ochmann J:
Dynamics of interleukin 1, 2, and 6 and tumor necrosis factor alpha
in multiple trauma patients. J Trauma. 36:336–340. 1994.
|
|
19
|
Majetschak M, Flach R, Heukamp T, et al:
Regulation of whole blood tumor necrosis factor production upon
endotoxin stimulation after severe blunt trauma. J Trauma.
43:880–887. 1997.
|
|
20
|
Spielmann S, Kerner T, Ahlers O, Keh D,
Gerlach M and Gerlach H: Early detection of increased tumour
necrosis factor alpha (TNFalpha) and soluble TNF receptor protein
plasma levels after trauma reveals associations with the clinical
course. Acta Anaesthesiol Scand. 45:364–370. 2001.
|
|
21
|
Zerey M, Burns JM, Kercher KW, Kuwada TS
and Heniford BT: Minimally invasive management of colon cancer.
Surg Innov. 13:5–15. 2006.
|
|
22
|
Feldman AM: TNF alpha - still a
therapeutic target. Clin Transl Sci. 1:1452008.
|
|
23
|
Kawasaki Y, Zhang L, Cheng JK and Ji RR:
Cytokine mechanisms of central sensitization: distinct and
overlapping role of interleukin-1beta, interleukin-6, and tumor
necrosis factor-alpha in regulating synaptic and neuronal activity
in the superficial spinal cord. J Neurosci. 28:5189–5194. 2008.
|
|
24
|
Oshima T, Lee S, Sato A, Oda S, Hirasawa H
and Yamashita T: TNF-alpha contributes to axonal sprouting and
functional recovery following traumatic brain injury. Brain Res.
1290:102–110. 2009.
|
|
25
|
Spinas GA, Keller U and Brockhaus M:
Release of soluble receptors for tumor necrosis factor (TNF) in
relation to circulating TNF during experimental endotoxinemia. J
Clin Invest. 90:533–536. 1992.
|
|
26
|
Van Zee KJ, Kohno T, Fischer E, Rock CS,
Moldawer LL and Lowry SF: Tumor necrosis factor soluble receptors
circulate during experimental and clinical inflammation and can
protect against excessive tumor necrosis factor alpha in vitro and
in vivo. Proc Natl Acad Sci USA. 89:4845–4849. 1992.
|
|
27
|
Gerlach H, Gerlach M and Clauss M:
Relevance of tumour necrosis factor-alpha and interleukin-1-alpha
in the pathogenesis of hypoxia-related organ failure. Eur J
Anaesthesiol. 10:273–285. 1993.
|
|
28
|
Rogy MA, Coyle SM, Oldenburg HS, et al:
Persistently elevated soluble tumor necrosis factor receptor and
interleukin-1 receptor antagonist levels in critically ill
patients. J Am Coll Surg. 178:132–138. 1994.
|
|
29
|
Pellegrini JD, Puyana JC, Lapchak PH,
Kodys K and Miller-Graziano CL: A membrane TNF-alpha/TNFR ratio
correlates to MODS score and mortality. Shock. 6:389–396. 1996.
|
|
30
|
Froon AH, Bemelmans MH, Greve JW, van der
Linden CJ and Buurman WA: Increased plasma concentrations of
soluble tumor necrosis factor receptors in sepsis syndrome:
correlation with plasma creatinine values. Crit Care Med.
22:803–809. 1994.
|
|
31
|
Balk RA: Pathogenesis and management of
multiple organ dysfunction or failure in severe sepsis and septic
shock. Crit Care Clin. 16:337–352. 2000.
|
|
32
|
Esposito E and Cuzzocrea S: Anti-TNF
therapy in the injured spinal cord. Trends Pharmacol Sci.
32:107–115. 2011.
|
|
33
|
Chio CC, Chang CH, Wang CC, et al:
Etanercept attenuates traumatic brain injury in rats by reducing
early microglial expression of tumor necrosis factor-α. BMC
Neurosci. 14:332013.
|
|
34
|
Shuh M, Bohorquez H, Loss GE Jr and Cohen
AJ: Tumor necrosis factor-α: life and death of hepatocytes during
liver ischemia/reperfusion injury. Ochsner J. 13:119–130. 2013.
|
|
35
|
Tobinick E, Kim NM, Reyzin G,
Rodriguez-Romanacce H and DePuy V: Selective TNF inhibition for
chronic stroke and traumatic brain injury: an observational study
involving 629 consecutive patients treated with perispinal
etanercept. CNS Drugs. 26:1051–1070. 2012.
|