Introduction
Gliomas are the most common type of brain tumor,
worldwide (1). Despite the use of
surgery, radiation and conventional chemotherapy, the median
survival time of patients with the most malignant type of glioma,
glioblastoma, is only one year (2).
Gliomas may be characterized by resistance to apoptosis and
radiation (3,4), however, the mechanism of this process
remains unclear.
According to previous studies, certain types of
glioma cells, including U87 cells, undergo senescence as opposed to
apoptosis following exposure to ionizing radiation (5–7).
Cellular senescence is an extremely stable form of cell cycle
arrest and constitutes a strong natural tumor suppressor mechanism.
At present, the predominant senescence signaling pathways
include the p53-p21Cip1/Waf1, p19Arf-Mdm2 and p16Ink4a-Rb pathways
(8).
B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1)
is a polycomb group protein that regulates cell proliferation and
has been found to be upregulated in a variety of human cancer
types, including acute myeloid leukemia and breast, colon, lung,
ovarian and nasopharyngeal cancer; this suggests a potential role
of Bmi-1 as an oncogene (9–12). Bmi-1 has been demonstrated to
regulate the expression of the p16Ink4a, hTERT and Hox group genes
(13). Overexpression of Bmi-1 may
reduce the expression of p16 and p19Arf (14,15),
which induce anti-senescence in tumor cells. Bmi-1 has also been
reported to be overexpressed in certain gliomas that usually
possess a poor prognosis (16–18).
The current study investigated the response of U87
glioma cells to radiation exposure as well as the role of Bmi-1 in
their response following radiotherapy.
Materials and methods
Cell culture
The human glioma cell line U87 was purchased from
the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
The glioma cells were maintained in minimum essential medium (GE
Healthcare Life Sciences, Logan, UT, USA) containing 10% fetal
bovine serum (FBS; GE Healthcare Life Sciences) and incubated at
37°C in a humidified atmosphere of 5% CO2.
Radiation
The cells were plated on 24-well dishes at a density
of 5×104 cells/0.5 ml on day 0. Subsequently, the U87
cells were immediately exposed to X-ray radiation with a linear
accelerator source (Elekta, Stockholm, Sweden) at a dose rate of
300 cGy/min. Every 24 h, the number of cells in three wells was
quantified using a cell counter (Inno-Alliance Biotech, Wilmington,
DE, USA) and the mean was calculated. The results are presented as
the mean ± standard error of three independent experiments.
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (/PI) double-labeled flow cytometry
(FCM)
To detect the apoptotic ratio of cells following 72
h of exposure to X-ray radiation, the expression of Annexin V-FITC
(Sigma-Aldrich, St. Louis, MO, USA) and the exclusion of PI
(Sigma-Aldrich) were detected by two-color FCM using the LSR
Fortessa cell analyzer (Becton-Dickinson, Franklin Lakes, NJ, USA).
The U87 cells were collected in Eppendorf PCR tubes, washed twice
with phosphate buffered saline (PBS) and resuspended in 500 μl
binding buffer. The samples were incubated with 5 μl Annexin V-FITC
at room temperature for 10 min and then 5 μl PI was added. Each
sample was incubated in the dark for a further 10 min at room
temperature prior to the fluorescence intensity being quantitated
by FCM.
Cell cycle
To detect the cell cycle of cells following 72 h of
exposure to radiation, glioma cells were fixed with 70% ethanol,
resuspended in PBS/1% FBS, and treated with ribonuclease (Beyotime
Institute of Biotechnology, Haimen, China). PI was added to the
cells and the samples were then analyzed by FCM (Becton-Dickinson).
Cell cycle profile analysis of the DNA histograms of integrated red
fluorescence was performed with ModFit LT 2.0 (Verity Software,
Inc., Topsham, ME, USA).
Senescence-associated β-galactosidase
(SA-β-Gal) staining
To detect the senescence ratio of cells following 72
h of exposure to radiation, SA-β-Gal staining was performed using
the SA-β-Gal Kit (Beyotime Institute of Biotechnology) following
the manufacturer’s instructions. The cells were considered to be
positive when the cytoplasm was stained with SA-β-Gal.
Western blot analysis
The U87 cells were collected and lysed in lysis
buffer (150 mM NaCl, 50 mM Tris with pH 7.4, 1% NP40, 0.1% SDS,
0.5% sodium deoxycholate), supplemented with protease inhibitors
(CWBio, Inc., Beijing, China) subsequent to 72 h of exposure to
radiation, which was followed by centrifugation at 10,000 × g for
15 min at 4°C. The protein concentration in each sample extract was
detected using the bicinchoninic acid assay (CWBio, Inc.). SDS-PAGE
was performed on 15% polyacrylamide gels, with 40 μg of protein
sample per lane. Following electrophoresis, the protein was
transferred to nitrocellulose membranes and incubated in 5% non-fat
milk at room temperature for 2 h. Subsequently, the membranes were
incubated overnight at 4°C with a primary polyclonal rabbit
anti-human antibody for Bmi-1 (sc-10745; Santa Cruz Biotechnology,
Dallas, TX, USA). Subsequent to being washed three times using PBS,
the membrane was incubated with an appropriate concentration of
horseradish peroxidase-conjugated anti-rabbit secondary antibody
(CWBio, Inc.) for 2 h. Following a further three washes with PBS,
the specific protein band was visualized using an enhanced
chemiluminescence kit (CWBio, Inc.).
Statistical analysis
Each experiment was conducted in triplicate.
Student’s T-test was used to evaluate the statistical significance
of the differences and a P<0.05 was considered to indicate a
statistically significant difference.
Results
Proliferation inhibition in U87 glioma
cells following X-ray radiation exposure
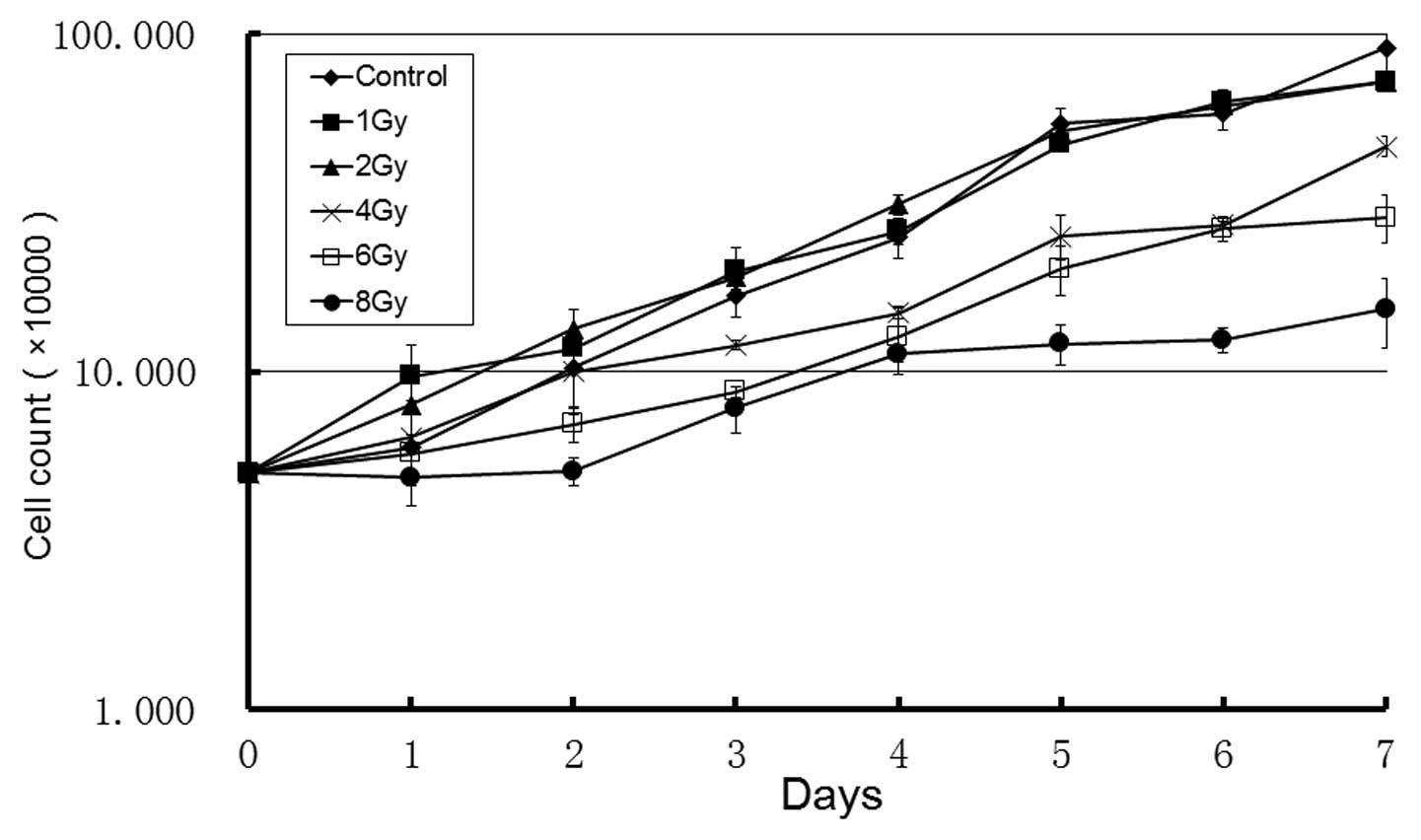
To determine the cellular proliferation of U87 cells
following exposure to radiation, the U87 cells were treated with
X-ray radiation at 1, 2, 4.6 and 8 Gy (Fig. 1). Cellular proliferation was
observed to be inhibited in a dose-dependent manner at doses ≥4 Gy.
No significant inhibition of cellular proliferation was observed in
the control, 1 and 2 Gy groups. Following 24 h of exposure to X-ray
radiation, the number of cells in the groups receiving doses of 1
and 2 Gy X-ray radiation was increased compared with the control
groups (P<0.05).
Apoptosis and senescence in U87 glioma
cells following exposure to X-ray radiation
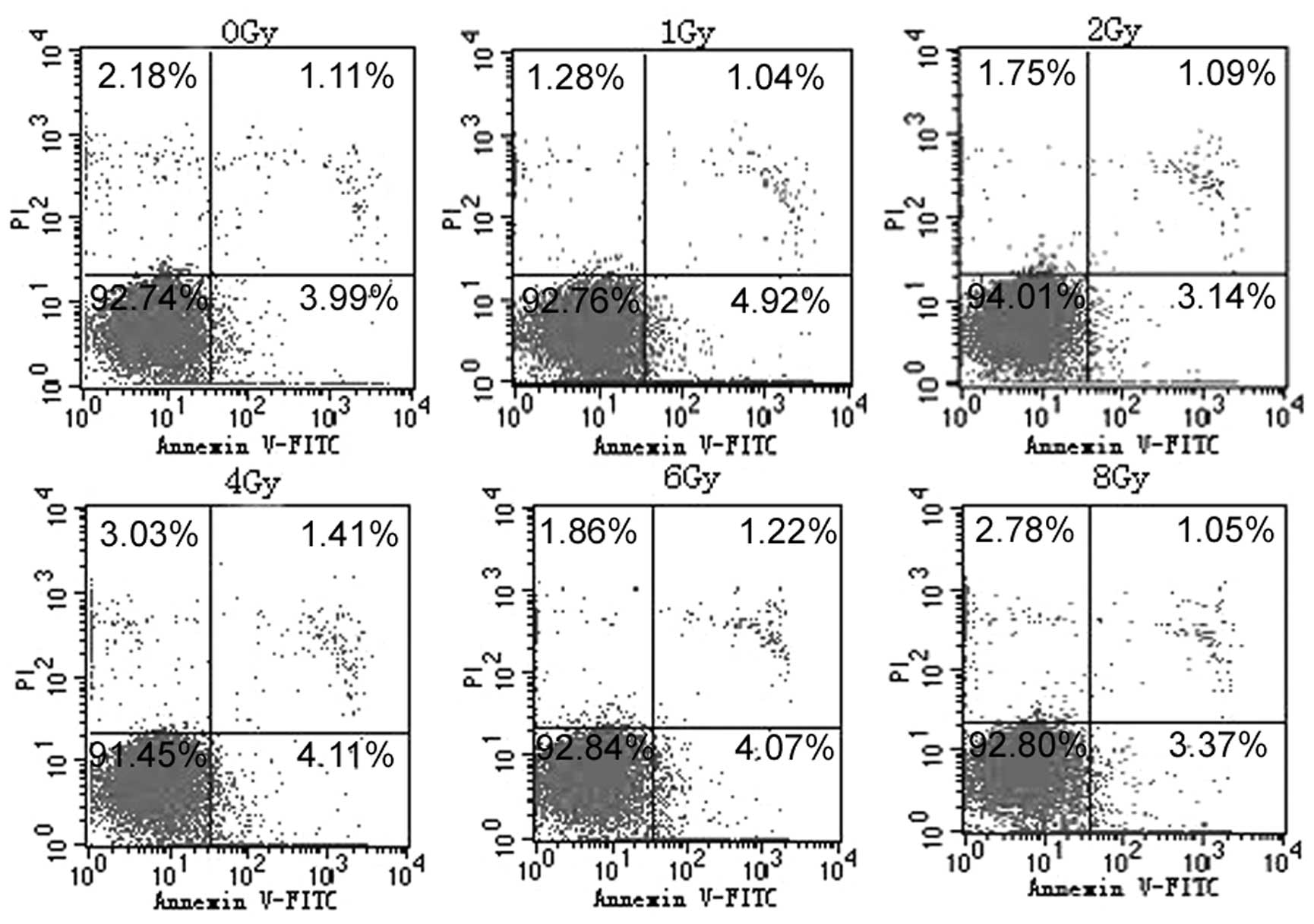
To evaluate whether the cellular proliferation
inhibition of U87 cells in response to X-ray radiation is
associated with apoptosis or senescence, U87 cells were treated
with X-ray radiation at 1, 2, 4.6 and 8 Gy. Following 72 h of
exposure to radiation, no significant apoptosis was identified in
any of the groups (Fig. 2). Using
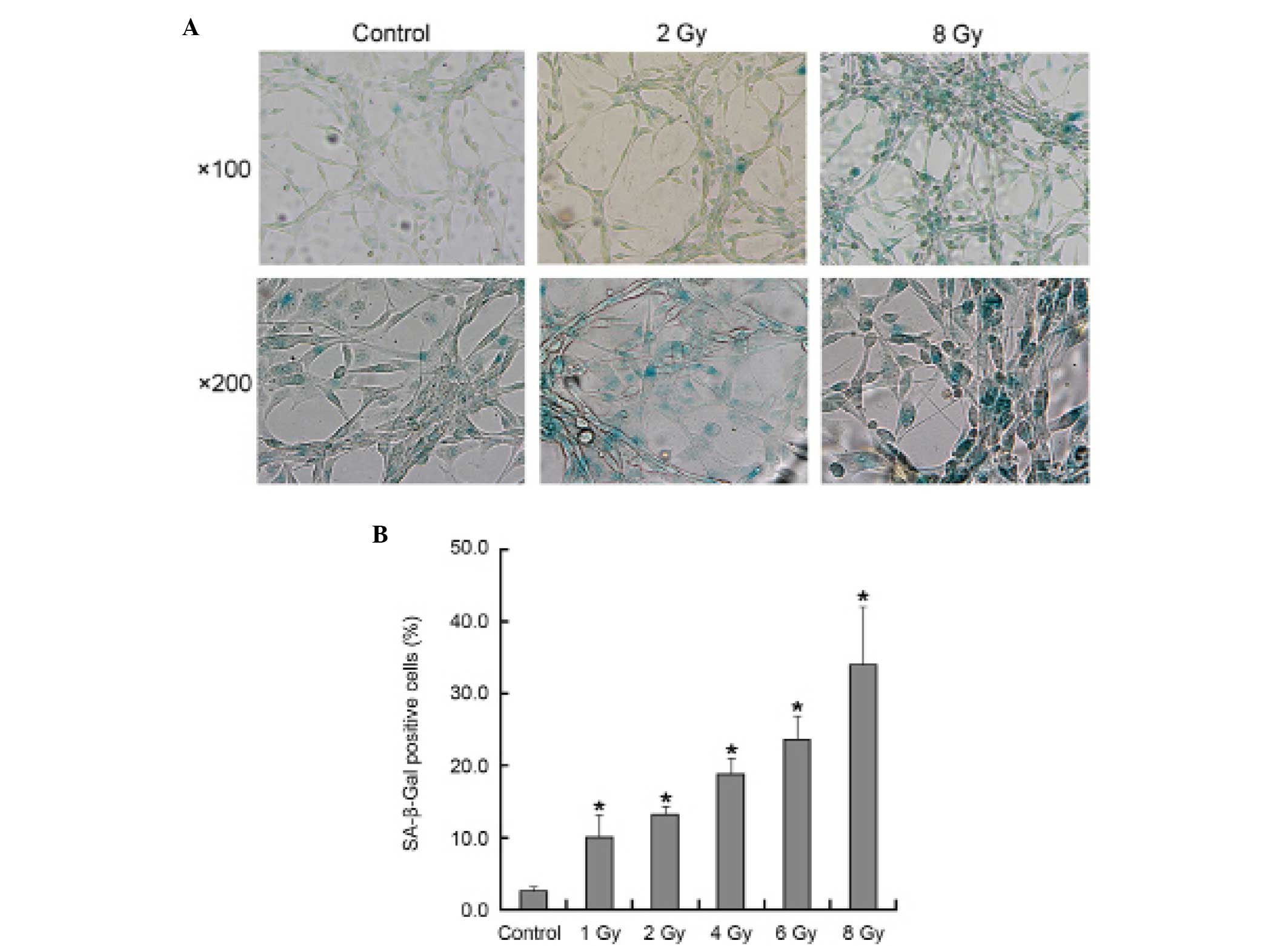
SA-β-Gal staining, it was observed that senescence had occurred in
all treatment groups in a dose-dependent manner (Fig. 3B). Under phase-contrast microscopy,
the senescent morphology of a large and flattened shape was
observed (Fig. 3A). These
morphological changes were more evident in the cells that were
exposed to radiation doses of ≥6 Gy.
The change of cell cycle phase in U87
glioma cells following exposure to X-ray radiation
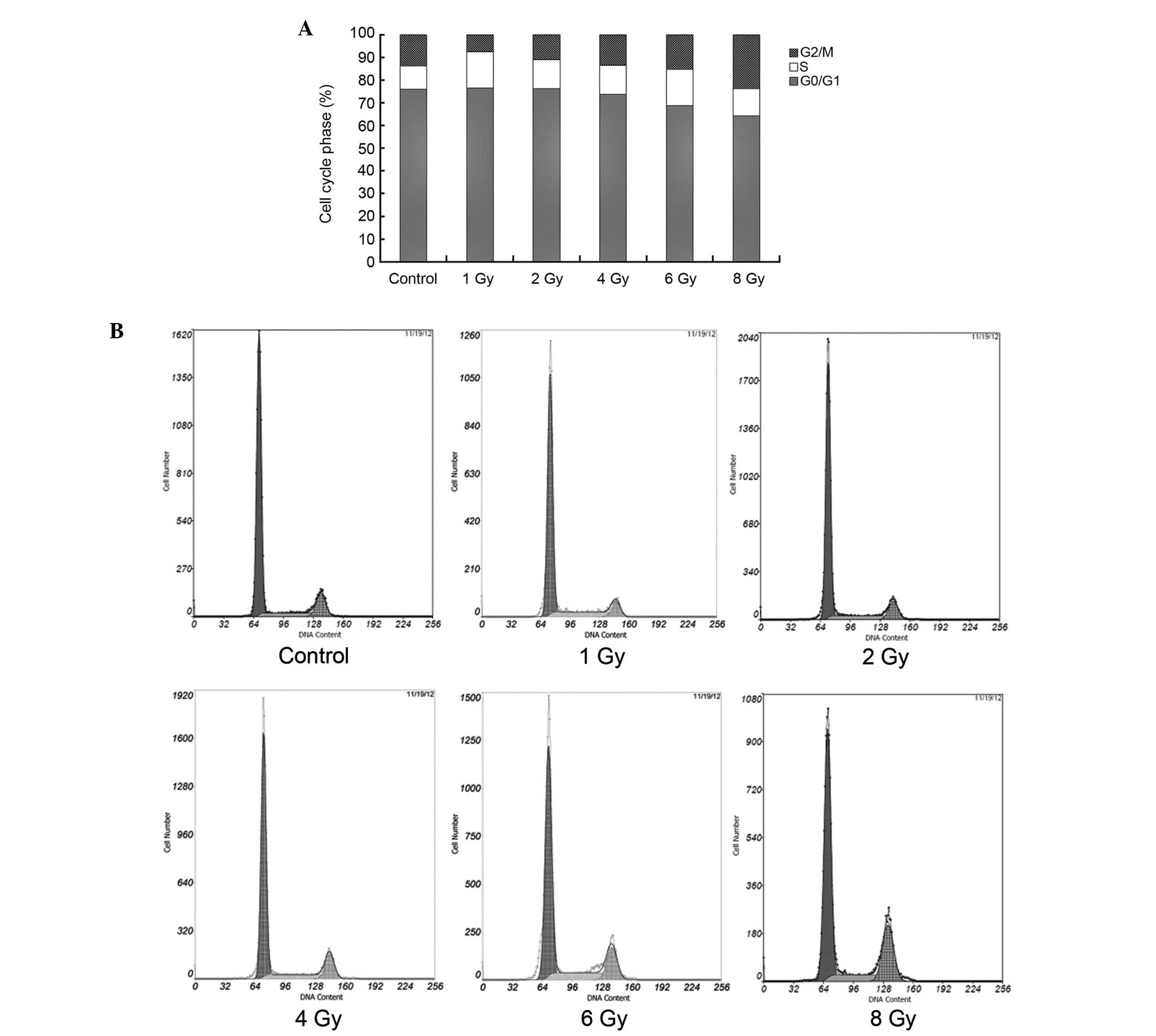
Following 72 h of exposure to X-ray radiation, the
cell cycle phases of U87 cells were analyzed (Fig. 4). The proportion of cells in the S
phase was increased in all of the treatment groups (P<0.05). The
proportion of cells in the G2/M phase was decreased in
the 1 and 2 Gy groups and increased in the 6 and 8 Gy groups
(P<0.05). No significant difference was identified in the
G0/G1 phase between the 1, 2, 4 Gy and
control groups, however, this was increased in the 6 and 8 Gy
groups.
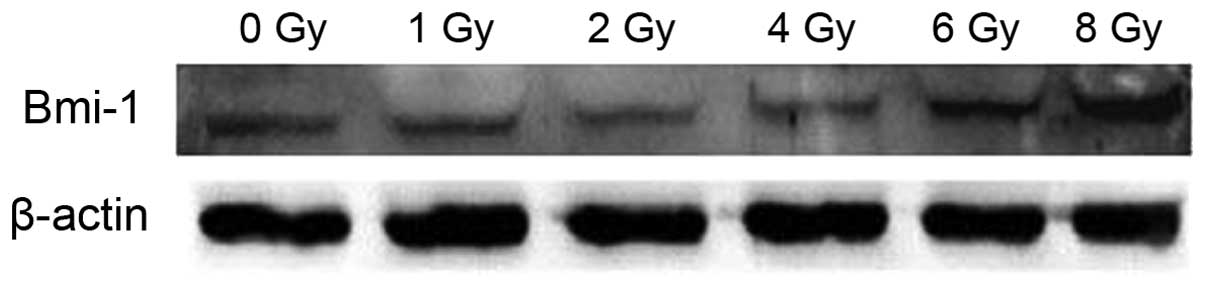
Expression of Bmi-1 in U87 glioma cells
following exposure to X-ray radiation
To observe whether the expression of Bmi-1 was
associated with exposure to X-ray radiation, U87 cells were exposed
to X-ray radiation in doses of 1, 2, 4.6 and 8 Gy. Following 72 h
of exposure to radiation, the expression of Bmi-1 was determined
(Fig. 5). Bmi-1 expression was
increased in the 6 Gy and 8 Gy groups (P<0.05). No significant
difference in Bmi-1 expression was observed between the 1, 2 and 4
Gy and control groups.
Discussion
Glioma is the most common malignant brain tumor in
adults, worldwide (19), with grade
IV glioblastoma being the most fatal (20,21).
Radiation is an important therapy for glioma patients, however, the
resistance of gliomas to radiation has affected the response to
treatment (22). If the mechanism
of radiation resistance is identified, novel treatment options for
radiation-resistant gliomas may be investigated to improve the
response to radiation treatment. U87 cells were selected for
investigation in the current study as they are a human glioblastoma
cell line that is characterized by resistance to apoptosis and
radiation.
Radiotherapy is significant in the treatment of
glioma. Numerous studies have focused on the more effective method
of increasing the radiosensitivity of glioma (23–25).
Radiation causes DNA damage to cells and subsequently induces
apoptosis, senescence or death. Apoptosis is known to be an
important mechanism for radiotherapy and chemotherapy, however, the
present study and others have demonstrated that radiation did not
induce apoptosis in certain gliomas, but instead induced senescence
(5,26,27).
Cellular senescence is characterized by irreversible
cell cycle arrest, with morphological changes that result in a
large and flattened cell shape. Senescent cells remain
metabolically active and exhibit certain properties, including
SA-β-Gal staining based on lysosomal β-galactosidase activity at pH
6.0, resistance to apoptosis and altered gene expression (28). Tumor cells have been hypothesized to
possess the ability to elude senescence. Therefore, it is possible
that improved tumor treatments may be developed by understanding
the underlying mechanism of senescence (29). The importance of cellular senescence
as a tumor suppression mechanism is increasingly being
recognized.
Bmi-1 may be significant in the senescence of tumor
cells (30). Bmi-1 negatively
controls the expression of the p16 gene (31,32);
p16 expression is one of the hallmarks of cellular senescence
(33) and inhibits the activity of
CDK4, which is the key enzyme in the G1-S transformation
during cell division, preventing the cells from entering S phase.
Once p16 is mutationally or transcriptionally inactivated in cells,
CDK4 is uncontrolled, leading to malignant proliferation. The tumor
suppressor gene, p16, is often mutationally and transcriptionally
inactivated in gliomas. In addition, Bmi-1 can induce the
coordination of the c-myc gene with p16 to promote cell
transformation and tumor formation, which results in the cell
escaping apoptosis (34). For these
reasons, Bmi-1 presents an attractive target for glioma
therapy.
The aim of the present study was to elucidate
whether Bmi-1 expression was associated with the senescence of the
U87 glioma cells that have been exposed to X-ray radiation. X-ray
radiation was unable to induce apoptosis in U87 glioma cells.
However, X-ray radiation was able to induce senescence, resulting
in the inhibition of U87 cell proliferation following exposure to
the radiation at a dose of ≥4 Gy. An increase in the proportion of
cells in the S phase was observed following exposure to various
doses of X-ray radiation. In addition, in the groups exposed to the
lower doses of 1,2 and 4 Gy, the proportion of cells in the
G2/M phase was decreased or not significantly different
compared with the control group. Furthermore, the proportion of
cells in the G0/G1 phase was not
significantly different compared with the control group. When cell
senescence is induced, the cell cycle should arrest in
G1 phase. This may be the reason that the proportion of
cells in the G0/G1 phase remained stable
despite the increase in the proportion of cells in the S phase.
However, in the higher dose group, exposed to 6 and 8 Gy, the
proportion of cells in the G2/M phase was increased and
the proportion of cells in the G0/G1 phase
was markedly decreased. Although the proportion of cells that were
SA-β-Gal positive was greater when compared with the control and
other radiation groups, which means that an increased number of U87
cells underwent senescence, the U87 cells did not arrest in the
G1 phase and continued to the S and G2/M
phases. It appeared that certain factors aid U87 cells in evading
senescence following exposure to higher doses of X-ray radiation.
In the present study, Bmi-1 expression was observed to be
significantly increased in the higher dose (6 and 8 Gy) groups and
therefore, may exert an effect that aids the cells in evading cell
cycle arrest. However, the mechanism associated with this progress
is not fully understood.
In the present study, senescence was determined as
the main mechanism of U87 cell growth suppression following
exposure to X-ray radiation. Bmi-1 may be significant in the
radioresistance of U87 cells by suppressing senescence. Future
studies focusing on the expression of Bmi-1 and its downstream
genes, using gene transfection technology in additional glioma cell
lines, may aid in determining the possible mechanism of Bmi-1 in
the radioresistance of glioma cells.
Acknowledgements
The present study was supported by Youth Foundation
in the Second Hospital of Shandong University (grant no.
Y2013010016), Jinan University Institutes Independent Innovation
Plan (grant no. 201401263), Development Project of Shandong
Province Science and Technology of Traditional Chinese Medicine
(grant no. 2007-140) and Shandong Province Natural Science
Foundation of China (grant no. 2012zre27087).
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, et al:
The 2007 WHO classification of tumours of the central nervous
system. Acta Neuropathol. 114:97–109. 2007.
|
|
2
|
Ohgaki H, Dessen P, Jourde B, et al:
Genetic pathways to glioblastoma: a population-based study. Cancer
Res. 64:6892–6899. 2004.
|
|
3
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008.
|
|
4
|
Ohgaki H and Kleihues P: Genetic
alterations and signaling pathways in the evolution of gliomas.
Cancer Sci. 100:2235–2241. 2009.
|
|
5
|
Lee JJ, Kim BC, Park MJ, et al: PTEN
status switches cell fate between premature senescence and
apoptosis in glioma exposed to ionizing radiation. Cell Death
Differ. 18:666–677. 2011.
|
|
6
|
Quick QA and Gewirtz DA: An accelerated
senescence response to radiation in wild-type p53 glioblastoma
multiforme cells. J Neurosurg. 105:111–118. 2006.
|
|
7
|
Nam HY, Han MW, Chang HW, et al:
Radioresistant cancer cells can be conditioned to enter senescence
by mTOR inhibition. Cancer Res. 73:4267–4277. 2013.
|
|
8
|
Hwang ES, Yoon G and Kang HT: A
comparative analysis of the cell biology of senescence and aging.
Cell Mol Life Sci. 66:2503–2524. 2009.
|
|
9
|
Chowdhury M, Mihara K, Yasunaga S, Ohtaki
M, Takihara Y and Kimura A: Expression of Polycomb-group (PcG)
protein BMI-1 predicts prognosis in patients with acute myeloid
leukemia. Leukemia. 21:1116–1122. 2007.
|
|
10
|
Vonlanthen S, Heighway J, Altermatt HJ, et
al: The Bmi-1 oncoprotein is differentially expressed in non-small
cell lung cancer and correlates with INK4A-ARF locus expression. Br
J Cancer. 84:1372–1376. 2001.
|
|
11
|
Zhang FB, Sui LH and Xin T: Correlation of
Bmi-1 expression and telomerase activity in human ovarian cancer.
Br J Biomed Sci. 65:172–177. 2008.
|
|
12
|
Song LB, Li J, Liao WT, et al: The
polycomb group protein Bmi-1 represses the tumor suppressor PTEN
and induces epithelial-mesenchymal transition in human
nasopharyngeal epithelial cells. J Clin Invest. 119:3626–3636.
2009.
|
|
13
|
Guo BH, Feng Y, Zhang R, et al: Bmi-1
promotes invasion and metastasis, and its elevated expression is
correlated with an advanced stage of breast cancer. Mol Cancer.
10:102011.
|
|
14
|
Dhawan S, Tschen SI and Bhushan A: Bmi-1
regulates the Ink4a/Arf locus to control pancreatic beta-cell
proliferation. Genes Dev. 23:906–911. 2009.
|
|
15
|
Bruggeman SW, Hulsman D, Tanger E, et al:
Bmi1 controls tumor development in an Ink4a/Arf-independent manner
in a mouse model for glioma. Cancer Cell. 12:328–341. 2007.
|
|
16
|
Glinsky GV, Berezovska O and Glinskii AB:
Microarray analysis identifies a death-from-cancer signature
predicting therapy failure in patients with multiple types of
cancer. J Clin Invest. 115:1503–1521. 2005.
|
|
17
|
Häyry V, Tynninen O, Haapasalo HK, et al:
Stem cell protein BMI-1 is an independent marker for poor prognosis
in oligodendroglial tumours. Neuropathol Appl Neurobiol.
34:555–563. 2008.
|
|
18
|
Li J, Gong LY, Song LB, et al: Oncoprotein
Bmi-1 renders apoptotic resistance to glioma cells through
activation of the IKK-nuclear factor-kappaB pathway. Am J Pathol.
176:699–709. 2010.
|
|
19
|
Taylor LP: Diagnosis, treatment, and
prognosis of glioma: five new things. Neurology. 75:S28–S32.
2010.
|
|
20
|
Stupp R, Mason WP, van den Bent MJ, et al;
European Organisation for Research and Treatment of Cancer Brain
Tumor and Radiotherapy Groups; National Cancer Institute of Canada
Clinical Trials Group. Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
|
|
21
|
Clarke J, Butowski N and Chang S: Recent
advances in therapy for glioblastoma. Arch Neurol. 67:279–283.
2010.
|
|
22
|
Stupp R, Hegi ME, Mason WP, et al;
European Organisation for Research and Treatment of Cancer Brain
Tumour and Radiation Oncology Groups; National Cancer Institute of
Canada Clinical Trials Group. Effects of radiotherapy with
concomitant and adjuvant temozolomide versus radiotherapy alone on
survival in glioblastoma in a randomised phase III study: 5-year
analysis of the EORTC-NCIC trial. Lancet Oncol. 10:459–466.
2009.
|
|
23
|
Xie C, Wang H, Cheng H, Li J, Wang Z and
Yue W: RAD18 mediates resistance to ionizing radiation in human
glioma cells. Biochem Biophys Res Commun. 445:263–268. 2014.
|
|
24
|
Liu P, Huang Z, Chen Z, et al: Silver
nanoparticles: a novel radiation sensitizer for glioma? Nanoscale.
5:11829–11836. 2013.
|
|
25
|
Naidu MD, Mason JM, Pica RV, Fung H and
Peña LA: Radiation resistance in glioma cells determined by DNA
damage repair activity of Ape1/Ref-1. J Radiat Res. 51:393–404.
2010.
|
|
26
|
Jendrossek V: The intrinsic apoptosis
pathways as a target in anticancer therapy. Curr Pharm Biotechnol.
13:1426–1438. 2012.
|
|
27
|
Dewey WC, Ling CC and Meyn RE:
Radiation-induced apoptosis: relevance to radiotherapy. Int J
Radiat Oncol Biol Phys. 33:781–796. 1995.
|
|
28
|
Schmitt CA: Cellular senescence and cancer
treatment. Biochim Biophys Acta. 1775:5–20. 2007.
|
|
29
|
Roninson IB: Tumor cell senescence in
cancer treatment. Cancer Res. 63:2705–2715. 2003.
|
|
30
|
Li Y, Hu J, Guan F, et al: Copper induces
cellular senescence in human glioblastoma multiforme cells through
downregulation of Bmi-1. Oncol Rep. 29:1805–1810. 2013.
|
|
31
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene Bmi-1
regulates cell proliferation and senescence through the ink4a
locus. Nature. 397:164–168. 1999.
|
|
32
|
Jiang L, Li J and Song L: Bmi-1, stem
cells and cancer. Acta Biochim Biophys Sin (Shanghai). 41:527–534.
2009.
|
|
33
|
Hara E, Smith R, Parry D, Tahara H, Stone
S and Peters G: Regulation of p16CDKN2 expression and its
implications for cell immortalization and senescence. Mol Cell
Biol. 16:859–867. 1996.
|
|
34
|
Molofsky AV, Pardal R, Iwashita T, Park
IK, Clarke MF and Morrison SJ: Bmi-1 dependence distinguishes
neural stem cell self-renewal from progenitor proliferation.
Nature. 425:962–967. 2003.
|