Introduction
A number of studies have demonstrated that apoptosis
is closely associated with the initiation, progression and
recurrence of cancer (1–3). Therefore, it is important to
manipulate apoptosis-regulating factors in the effective treatment
of cancer patients. Human programmed cell death 5 (PDCD5), formerly
designated as TF-1 cell apoptosis-related gene 19, was cloned from
TF-1 cells during the apoptotic process induced by cytokine
withdrawal. PDCD5 plays a significant role in cellular apoptosis,
and its overexpression in TF-1, MGC-803 and HeLa cells facilitates
apoptosis triggered by growth factor or serum withdrawal (4). PDCD5 is widely expressed in various
tissues and its mRNA expression level is significantly higher in
adult tissues than in fetal tissues. In cells undergoing apoptosis,
the level of PDCD5 protein is significantly increased and is
located in the nuclei preceding the externalization of
phosphatidylserine and the fragmentation of chromosomal DNA
(5).
The decreased expression of PDCD5 has been reported
in various human tumors, including prostate (6), lung (7) and ovarian (8) cancer, gliomas (9) and leukemia (10). PDCD5 also enhances apoptosis by
cooperating with cisplatin [cis-diamminedichloroplatinum(II)] in
certain tumor cells, including those of colorectal cancer, gastric
cancer and glioma (11–13).
Cisplatin is a common drug for cancer chemotherapy,
however, with repeated exposure, tumor cells often become resistant
to its effects. Hepatocellular carcinoma (HCC) is known to be
resistant to various chemotherapeutics, including cisplatin.
Studies have shown that this resistance is likely to be
attributable to the cisplatin-induced upregulation of hTERT and the
PI3K-dependent survivin pathway (14,15).
Therefore, we hypothesize that the exogenous overexpression of
PDCD5 may enhance apoptosis and reverse cisplatin resistance in
HCC. In the present study, the biological behavior of HCC cells
were analyzed in vitro by the stable transfection of the
PDCD5 gene, and the effects on apoptosis induced by cisplatin and
invasion by transforming growth factor (TGF)-β were
investigated.
Materials and methods
Cell culture
The human HCC cell line, HepG2, was purchased from
the Institute of Biochemistry and Cell Biology, Shanghai Institutes
for Biological Sciences, Chinese Academy of Sciences (Shanghai,
China). The cells were incubated in complete Dulbecco’s modified
Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented
with 10% heat-inactivated fetal bovine serum (FBS; Sijichun
Bioengineering Materials Inc., Hangzhou, Zhejiang, China), 100 U/ml
penicillin and 100 μg/ml streptomycin, in a humidified incubator at
37°C with 5% CO2.
Construction and transfection of PDCD5
plasmid
A PDCD5 full length cDNA sequence was obtained from
GenBank (http://www.ncbi.nlm.nih.gov/genbank/; accession
number, NM_004708.3). Total RNA was extracted using oligo (dT) from
the human HCC HepG2 cells and was reverse transcribed as a template
for reverse transcription polymerase chain reaction (RT-PCR). The
primer sequences were as the follows: Sense, 5′-CGC GGA TCC CCG AGG
GGC TGC GAG AGT GA-3′ and antisense, 5′-CGC GAA TTC CCT AGA CTT GTT
CCG TTA AG-3′. PCR conditions of 40 cycles of 94°C for 30 sec, 60°C
for 45 sec and 72°C for 30 sec followed by a final elongation step
at 72°C for 10 min, were used. The PCR products of full-length
PDCD5 cDNA were then ligated into the BamHI/EcoRI
sites of the eukaryotic expression vector, pcDNA3.1/Neo(+)
(Invitrogen) using T4 DNA ligase at 16°C overnight, followed by
transformation of competent Escherichia coli DH5α. DNA
sequencing was used to identify a recombinant plasmid clone with
the correct sequence, and this bacterial clone was amplified and
purified in for eukaryote transfection. The HepG2 cells were
transfected with pcDNA3.1-PDCD5 plasmid or pcDNA3.1-Neo plasmid
(empty vector) [pcDNA3.1(+)] using Lipofectamine™ 2000 (Invitrogen,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
RT-PCR and RT-quantitative (q)PCR were performed to detect PDCD5
mRNA expression 48 h after transfection. SuccessfulLY transfected
HepG2 cells were then grown in complete medium for further G418
screening (400 μg/ml; Sigma-Aldrich, St. Louis, MO, USA). After
four weeks, colonies were isolated and expanded into cell clones.
The subclone cells expressing only Neo or Neo and PDCD5 genes were
termed HepG2-Neo and HepG2-PDCD5, respectively.
RT-PCR analysis
The levels of PDCD5 mRNA were first examined by
RT-PCR and β-actin was used as an internal reference. Total RNA (5
μg) was isolated from the HepG2 cells 48 h after transfection and
RT was performed to synthesize cDNA using random primers with
Easyscript First-Strand cDNA Synthesis SuperMix (TransGen Biotech,
Beijing, China) primed with oligo(dT18). The forward and
reverse primers were synthesized by Sangon Biotech (Shanghai) Co.,
Ltd., (Beijing, China), and the sequences and expected sizes of the
PCR products were as follows: PDCD5 forward, 5′-ACA GAT GGC AAG ATA
TGG ACA-3′ and reverse, 5′-TCC TAG ACT TGT TCC GTT AAG-3′ (210bp);
and β-actin forward, 5-CGG GAA ATC GTG CGT GAC ATT-3′ and reverse,
5′-CTA GAA GCA TTT GCG GTG GAC-3′ (510bp). The thermal procedure of
PCR for PDCD5 and β-actin mRNA was performed at 94°C for 4 min for
1 cycle, then 94°C for 45 sec, 52°C for 45 sec and 72°C for 1 min
for 30 cycles, and 72°C for 7 min for 1 cycle. PCR products were
subjected to electrophoresis on 1.5% agarose gels containing
ethidium bromide and then visualized under ultraviolet light.
qPCR analysis
To quantify the results of the RT-PCR, PDCD5 mRNA
expression levels were further analyzed by RT-qPCR analysis, which
was performed by an RT-Cycler™ Real Time PCR Detection System
(CapitalBio, Ltd., Beijing, China) with SYBR Green (Molecular
Probes, Invitrogen). The following primers were used: Sense, 5′-ACA
GAT GGC AAG ATA TGG ACA-3′ and anti-sense, 5′-TCC TAG ACT TGT TCC
GTT AAG-3′ (199 bp) for PDCD5 and; sense, 5′-TTA GTT GCG TTA CAC
CCT TTC-3′ and anti-sense, 5′-ACC TTC ACC GTT CCA GTT T-3′ (150 bp)
for β-actin. Firstly, RNA was extracted and reverse transcribed to
cDNA using Easyscript First-Strand cDNA Synthesis SuperMix (Beijing
TransGen Biotech Co., Ltd., Beijing, China), then 1 μl cDNA was
added in a 20-μl reaction mixture containing 0.5X SYBR Green, 1X
TransStart Green qPCR Supermix (Transgen Biotech) and 0.5 μmol/l
primer sets. The cycling conditions were as follows: 95°C for 5 min
for 1 cycle, followed by 95°C for 45 sec, 57°C for 20 sec and 72°C
for 20 sec for 40 cycles. The expression levels of PDCD5 were
internally normalized to β-actin. The relative expression level of
PDCD5 mRNA was calculated by the 2−ΔΔCT method. Each
experiment was performed in duplicate and repeated three times.
Cell viability assay
The cell growth rate was determined by MTT assay
(Sigma-Aldrich). Briefly, cells (100 μl) at the logarithmic growth
phase were seeded at a 1×104/ml density into 96-well
culture plates. MTT solution (10 μl; 5 mg/ml) was added into each
well and incubated at 37°C for 4 h. Following centrifugation at
1,409 × g for 10 min, the supernatant was discarded and 100 μl DMSO
was added. When the remaining formazan pellet was dissolved
completely, the absorbance values at a 570 nm wavelength were read
on an ELISA plate reader (Bio-Rad, Hercules, CA, USA). The total
procedure was repeated three times.
Flow cytometry analysis of the cell
cycle
HepG2 cells at the logarithmic growth phase were
seeded in 6-well plates. After reaching 50% confluence, the
adherent cells were cultured in serum-free medium for 24 h and then
were cultured in DMEM supplemented with 10% FBS. After 48 h, the
cells were digested and harvested with 250 μl trypsin. The cell
pellet was obtained following centrifugation for 3 min at 4°C and
978 × g, and was re-suspended with 300 μl ice-cold PBS on ice,
followed by resuspension in 70% ethanol at 4°C for 30 min. Finally,
1 ml propidium iodide (PI) staining solution (20 μg/ml PI, 0.1%
Triton X-100, 2 mM EDTA and 8 μg/ml DNase-free RNase) was added to
the samples, and then the associated data were analyzed on a
FACScan (Becton-Dickinson, San Francisco, CA, USA). Results were
acquired from 10,000 cells.
Flow cytometric analysis of
apoptosis
Next, an Annexin V-fluorescein isothiocyanate (FITC)
apoptosis detection kit (BD Biosciences Clontech, California) was
used to identify the translocation of phosphatidylserine. The HepG2
cells were cultured with 5 μg/ml cisplatin. After 24 h,
2×105 cells in each well were harvested. Subsequent to
being washed with PBS buffer, the cell pellet was incubated with
2.5 μl Annexin V and 5 μl PI (final concentration, 10 μg/ml) in 100
μl 1X binding buffer for 15 min in the dark. Apoptosis was
determined by flow cytometry and analyzed using CellQuest and
Modfit software (Becton-Dickinson). At least 10,000 events were
analyzed for each sample.
Cell migration assay
The Boyden chamber invasion assay was performed to
evaluate the in vitro migration of the HepG2-Neo and
HepG2-PDCD5 cells in a 24-well tissue culture plate with a
Transwell filter membrane. The lower side of the filters was coated
with type I collagen (0.5 mg/ml). The cells were seeded in the
upper part of the Transwell plate at a density of
5×105/ml, with 100 μl cell suspension in each well.
After 24 h, the cells on the upper surface of the filter were
removed, and the remaining cells were fixed with methanol and
stained with hematoxylin and eosin (Sigma-Aldrich). Cell counting
was performed under light microscopy (x200 magnification) and the
cells that had migrated to the lower chamber were regarded as
migrated cells. Each sample was assayed in triplicate and repeated
twice
Western blot analysis
Proteins from the HepG2 cells were extracted and
their concentrations were determined by bicinchoninic acid protein
concentration assay kit (Beijing Biosea Biotechnology, Co., Ltd.,
Beijing, China). The cell lysates (50 μg) were resolved on 15%
SDS-polyacrylamide gels, electrophoretically transferred to
polyvinylidene difluoride membranes and then incubated with primary
monoclonal mouse antibodies against PDCD5, phospho-p53, Snail or
insulin-like growth factor (IGF)-1 (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). The horseradish peroxidase-conjugated rabbit
anti-mouse secondary antibody was used at 1:1,000 dilutions for 2 h
at room temperature. Blots were visualized using the
chemiluminescence method. β-actin was used as an internal
control.
Statistical analysis
All quantitative data are expressed as the mean ±
standard deviation. The statistical analysis was performed by
commercially available SPSS 14.0 software (SPSS, Inc., Chicago, IL,
USA). Student’s t-test (unpaired, two-tailed) was performed to
compare the means between two groups. The means of the different
groups were compared using a one-way analysis of variance.
P<0.05 was used to indicate a statistically significant
difference.
Results
Construction of eukaryotic expression
vector of pcDNA3.1/Neo(+)-PDCD5
Following the DNA sequencing analysis for the
validation of recombinant pcDNA3.1/Neo(+)-PDCD5 (data not shown),
G418 screening was performed to select for cells with successful
transfection. All untransfected HepG2 cells were dead following
G418 (400 μg/ml) selection. The pcDNA3.1/Neo(+)-PDCD5 transfected
cells were cultivated with G418 for four weeks, until viable clones
could be observed. The clones were selected for further
amplification. The RT-PCR, RT-qPCR and western blot analyses were
performed to measure the mRNA and protein expression levels of
PDCD5 in the HepG2, HepG2-Neo and HepG2-PDCD5 cells. The mRNA and
protein levels of PDCD5 in the HepG2-PDCD5 cells were significantly
higher than those in the HepG2 and HepG2-Neo cells (P<0.05). No
significant difference was found in PDCD5 expression between the
HepG2 and HepG2-Neo cells (P>0.05) (Fig. 1).
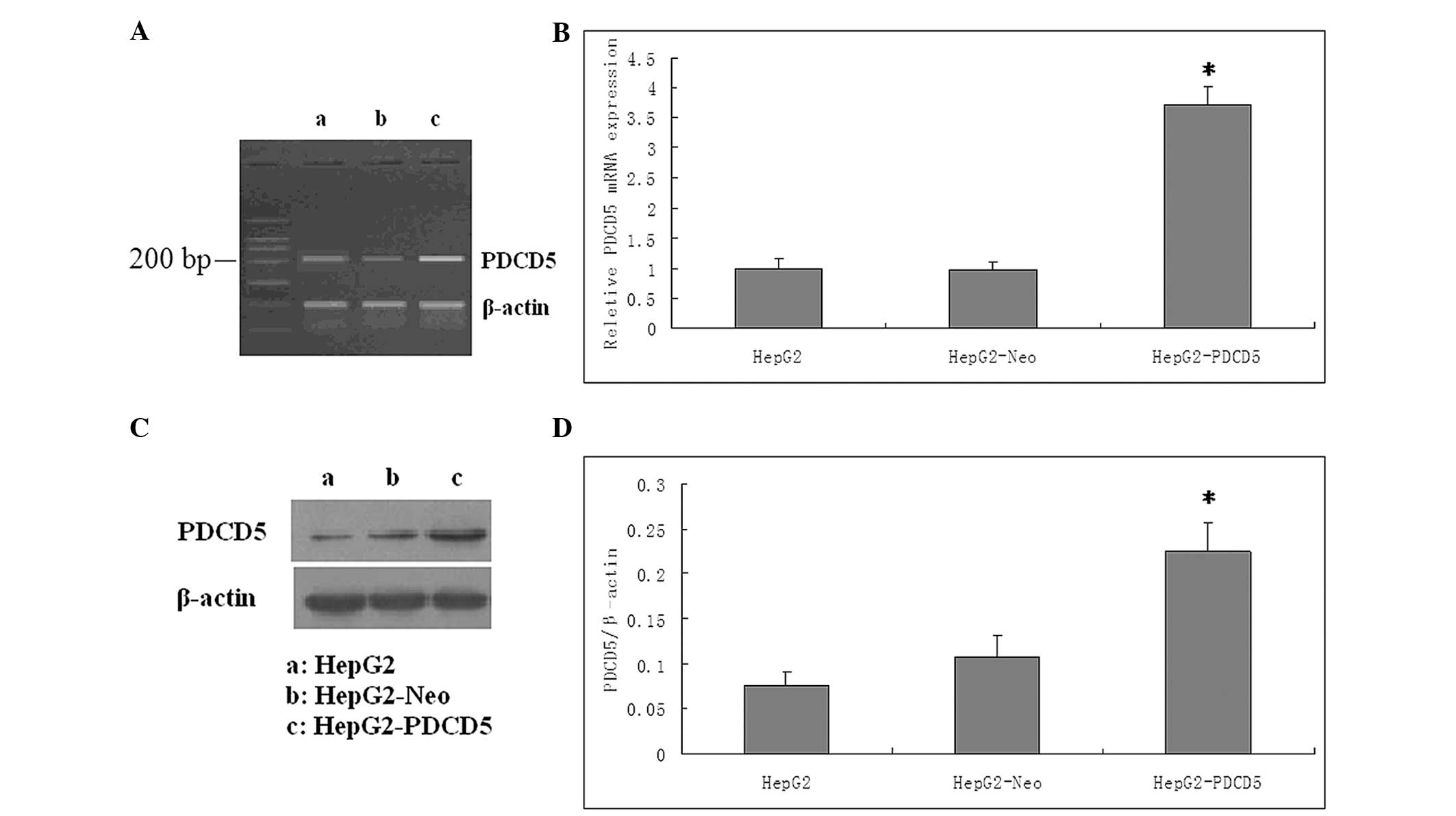 | Figure 1Validation for the efficacy of PDCD5
overexpression at the mRNA and protein levels. (A) The PDCD5 mRNA
expression level was markedly increased by pcDNA3.1-PDCD5 plasmid
transfection, as shown by the reverse transcription polymerase
chain reaction (RT-PCR) products in the agarose gel. a, HepG2
cells; b, HepG2-Neo cells; c, HepG2-PDCD5 cells (B) Expression of
PDCD5 mRNA was detected by RT-quantitative PCR analysis. The level
of PDCD5 mRNA following transfection was increased by three-fold
(P<0.05). The control plasmid had no effect on the PDCD5 mRNA
expression level in the HepG2 cells. (C) PDCD5 protein expression
was detected by western blot analysis. PDCD5 protein expression was
higher in the cells transfected with PDCD5 than in the wild-type
cells or cells transfected with control plasmid. β-actin served as
a loading control. One representative figure is shown from three
independent experiments. a, HepG2 cells; b, HepG2-Neo cells; c,
HepG2-PDCD5 cells (D) Relative expression of PDCD5 of the three
groups. The Y axis indicates the gray value of PDCD5 normalized to
that of β-actin. Data are expressed as the mean ± standard
deviation. A two-tailed, unpaired t-test was performed.
*P<0.05 vs. HepG2 group (n=6). PDCD5, programmed cell
death 5; HepG2-Neo, HepG2 cells transfected with control plamid;
HepG2-PDCD5, HepG2 cells transfected with PDCD5 plamid. |
Changes of the biological behavior of the
tumor by the stable transfection of PDCD5
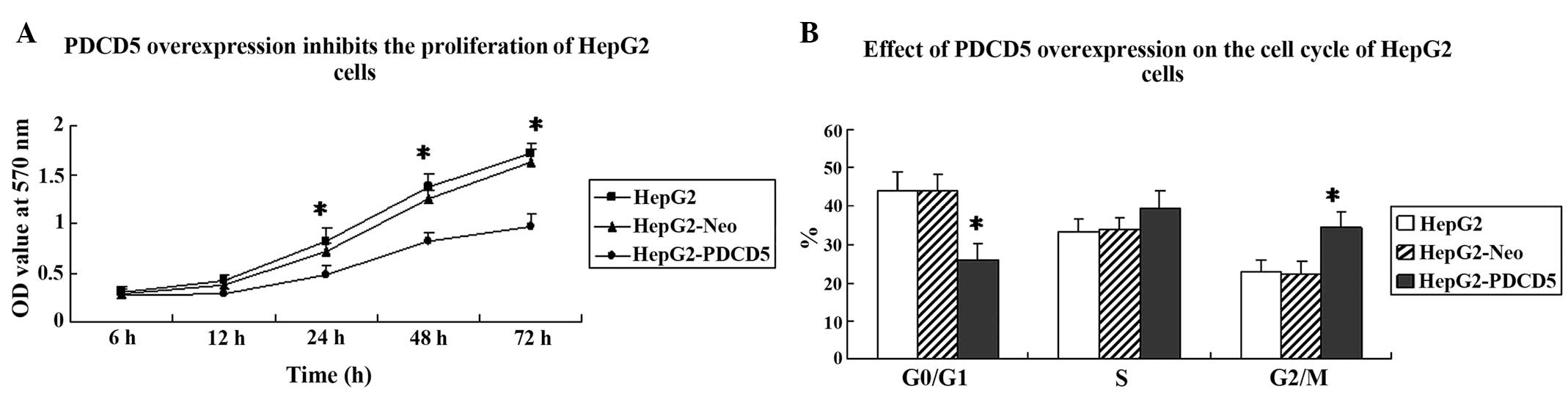
To analyze the effect of PDCD5 overexpression on
cancer cell growth, an MTT assay and flow cytometry were performed
to assess cell proliferation and the cell cycle. Cell proliferation
was significantly slower in the HepG2-PDCD5 cells compared with the
HepG2 and HepG2-Neo cells (Fig.
2A). Next, an analysis of the cell cycle was performed on the
HepG2, HepG2-Neo and HepG2-PDCD5 cells. The percentage of cells in
the G2/M phase was significantly higher in the
HepG2-PDCD5 cells compared with the HepG2 and HepG2-Neo cells
(P<0.05) (Fig. 2B). This
indicated that PDCD5 overexpression induces G2/M cell
cycle arrest in HepG2 cells.
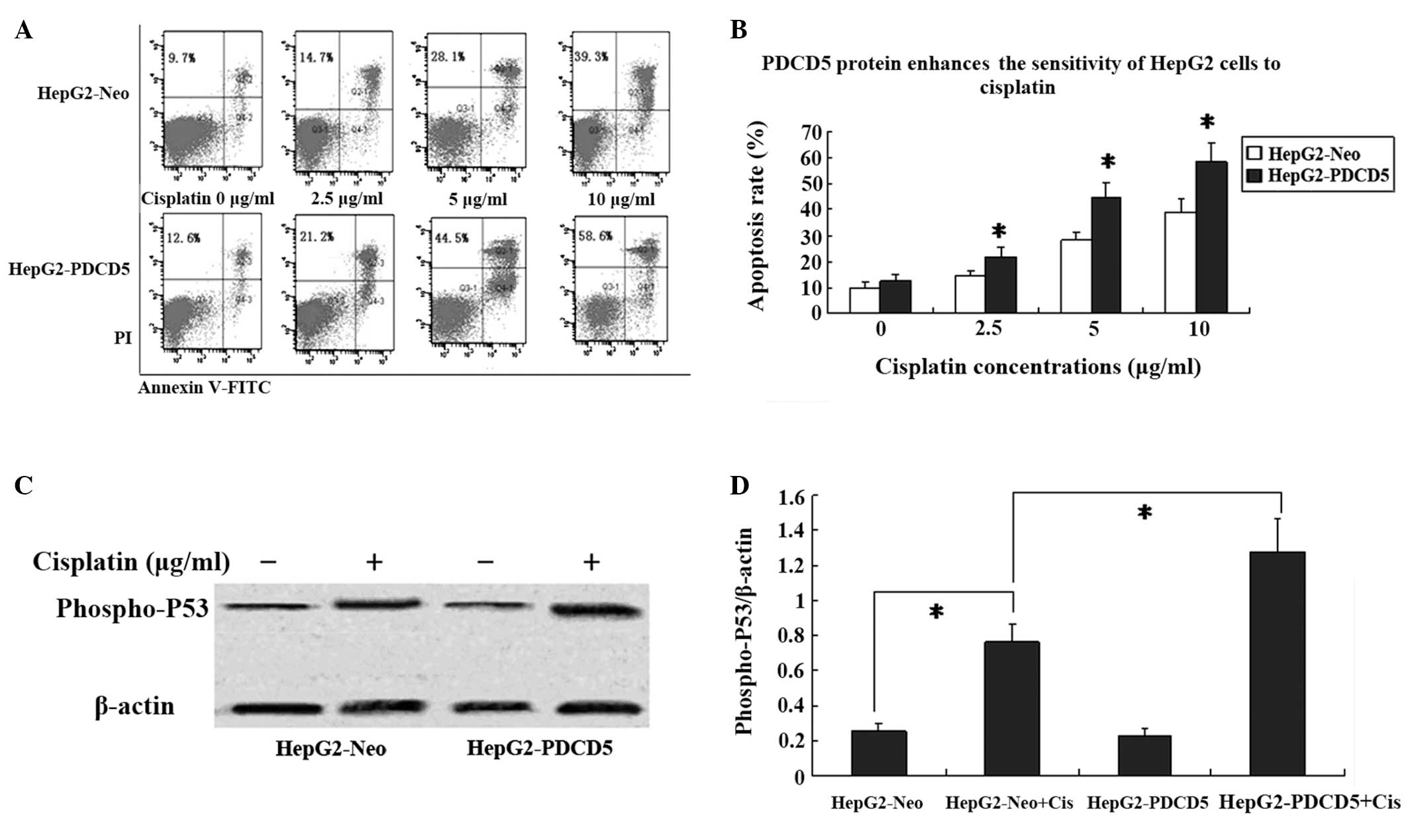
PDCD5 protein enhances the sensitivity of
HepG2 cells to cisplatin in vitro
The HepG2-Neo and HepG2-PDCD5 cells were treated
with varying concentrations of cisplatin and the chemotherapeutic
sensitivity was assessed by Annexin V-FITC and PI double staining.
Following treatment with 2.5, 5 and 10 μg/ml cisplatin for 24 h,
the number of HepG2-Neo and HepG2-PDCD5 apoptotic cells were
increased in a dose-dependent manner (Fig. 3A and B). In the cells without
cisplatin treatment, no difference was found in the apoptotic rate
between the HepG2-Neo and HepG2-PDCD5 cells.
PDCD5 interacts with p53 proteins in a variety of
cancer cells. To assess the effect of PDCD5 on p53 protein, the
level of phospho-p53 protein was measured by western blot analysis.
Phospho-p53 protein expression was increased following 24 h of
treatment with cisplatin in the HepG2-Neo and HepG2-PDCD5 cells.
Moreover, the HepG2-PDCD5 cells showed higher phospho-p53 protein
levels compared with the HepG2-Neo cells following cisplatin
treatment (Fig. 3C and D). No
difference was found in the p53 protein expression level between
the HepG2-Neo and HepG2-PDCD5 cells without cisplatin
treatment.
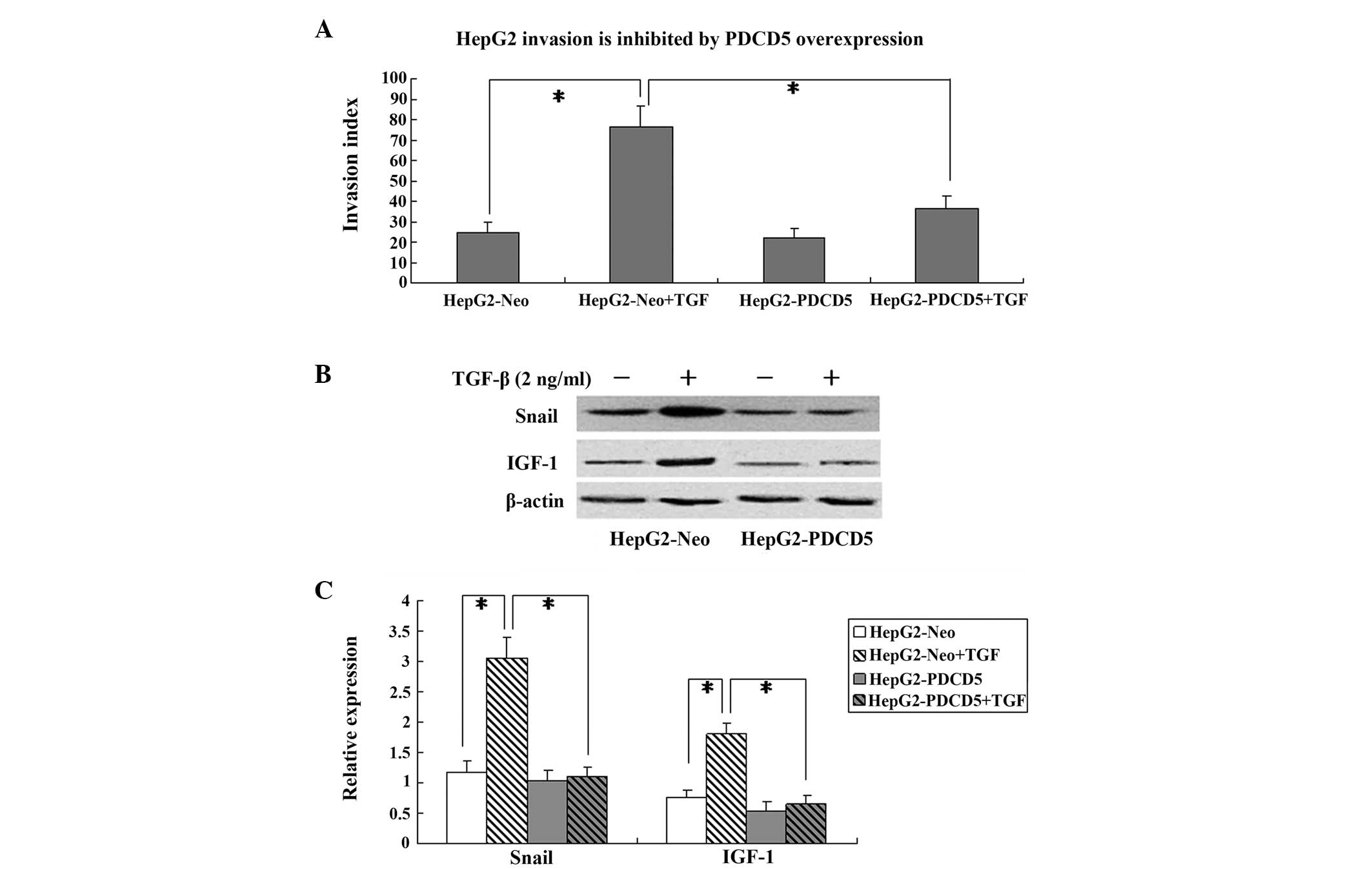
PDCD5 overexpression reduces invasion and
epithelial-mesenchymal transition (EMT) induced by TGF-β
The HepG2-Neo and HepG2-PDCD5 cells were incubated
with TGF-β (2 ng/ml) for 24 h to induce invasion and EMT. The
Boyden chamber invasion assay showed that PDCD5 transfection
exhibited no effect on the invasion index in the cells without
TGF-β treatment. However, in the TGF-β-treated HepG2 cells, PDCD5
transfection significantly decreased the invasion index compared
with the cells transfected with the control plasmid (P<0.05)
(Fig. 4A).
The expression of the EMT marker, Snail, was
determined by western blotting. In the cells transfected with the
HepG2-Neo control plasmid, Snail protein was significantly
increased by TGF-β treatment. However, PDCD5 transfection
significantly decreased Snail protein expression in the HepG2 cells
treated with TGF-β (P<0.05) (Fig. 4B
and C).
IGF-1 protein expression was also measured in the
HepG2 cells by western blotting. TGF-β treatment significantly
increased the IGF-1 protein expression level in the HepG2-Neo
cells, which was attenuated in the HepG2-PDCD5 cells (Fig. 4B and C). This indicates that PDCD5
may downregulate IGF-1 protein expression in TGF-β-treated HepG2
cells.
Discussion
In the present study, the PDCD5 gene was stably
transfected into a human HCC cell line to induce its
overexpression. It was demonstrated that the transfection of PDCD5
into HepG2 cells could change the biological behaviors of tumors,
such as the cellular proliferation, cell cycle progression,
cisplatin sensitivity, tumor invasion and EMT. The growth of the
HepG2-PDCD5 cells was slower than that of the HepG2 and HepG2-Neo
cells. A higher percentage of HepG2-PDCD5 cells were in the
G2/M phase compared with the HepG2 and HepG2-Neo cells.
The PDCD5-transfected cells showed higher sensitivity to cisplatin
treatment compared with the HepG2-Neo cells, with higher p53
protein expression. PDCD5 overexpression can attenuate tumor
invasion, EMT and IGF-1 protein induced by TGF-β treatment.
In the present study, the HepG2-PDCD5 cells had
decreased levels of proliferation compared with the HepG2 and
HepG2-Neo cells, as demonstrated by the presence of less viable
cells. This indicated that PDCD5 may participate in the
pathogenesis of tumors, and will be associated with various
clinicopathological factors in HCC patients. The decreased
expression level of PDCD5 has been observed in various human
tumors, and is correlated with high-grade astrocytic gliomas
(9), a higher Gleason grade in
prostate cancer (6) and an advanced
International Federation of Gynecologists and Obstetricians stage
and poorer survival in epithelial ovarian carcinomas (8). To the best of our knowledge, no study
has been reported on the correlation between PDCD5 expression and
the clinicopathological factors in HCC patients; this requires
further investigation.
The present study found that the stable transfection
of PDCD5 could also induce cell cycle arrest, as demonstrated by
the higher percentage of HepG2-PDCD5 cells in the G2/M
phase compared with the HepG2 and HepG2-Neo cells. This suggests
that the decreased number of viable HepG2-PDCD5 cells is partly
caused by the inhibition of cell cycle progression. G2/M
is an important cell cycle checkpoint prior to cells entering the
mitotic phase. In the present study, G2/M arrest
following PDCD5 transfection depended on functional p53, since in
cells with inactive p53, G2/M arrest did not occur
(16,17). This indicates that in HepG2 cells,
p53 protein is intact and cisplatin resistance could be reversed
(18). Therefore, the present study
evaluated whether PDCD5 overexpression enhances the sensitivity of
HepG2 cells to cisplatin in vitro.
Cisplatin is a second-generation platinum-based
chemotherapeutic drug for the clinical treatment of a variety of
tumors. Cisplatin functions through restraining DNA replication,
leading to cell cycle arrest and apoptosis. The long-term use of
cisplatin results the development of drug resistance in numerous
patients with HCC, leading to disease recurrence (19). Therefore, increasing the sensitivity
of HCC cells to cisplatin may aid in overcoming drug resistance and
tumor recurrence. The present results demonstrated that HepG2 cells
with stable transfection of PDCD5 exhibited an enhanced sensitivity
to cisplatin (5 μg/ml) exposure, as demonstrated by the higher
apoptosis rates compared with the HepG2-Neo cells. The
aforementioned results showed that PDCD5 can induce G2/M
phase cell cycle arrest in HepG2 cells, and indicates functional
p53 protein within the cells. Therefore, the study investigated
whether p53 is able to participate in the enhanced sensitivity to
cisplatin caused by PDCD5. The results showed that higher
phospho-p53 protein levels were observed in the HepG2-PDCD5 cells
compared with the HepG2-Neo cells following cisplatin treatment,
and suggests that elevated phospho-p53 protein levels may mediate
enhanced apoptosis by PDCD5 transfection following cisplatin
treatment. In fact, PDCD5 can directly bind with p53 protein
(20), and co-localization of p53
and PDCD5 protein has been found in the synergistic therapeutic
effect of PDCD5 with cisplatin (12). The present results were further
supported by a recent study that showed that the knockdown of PDCD5
by RNA interference decreased the level of p53 phosphorylation
(21). This indicates that PDCD5
may function as a co-activator of p53 upon DNA damage, such as that
caused by cisplatin treatment.
The present study also found that PDCD5
overexpression could reduce invasion and EMT induced by TGF-β.
Tumor invasion is a complex and multi-step process that involves
alteration of the cell adhesion to extracellular matrix proteins.
TGF-β has been shown to promote vascular invasion in HCC (22). The present results showed that the
pro-invasive effect of TGF-β was reversed by PDCD5 overexpression.
There are few studies on the correlation between PDCD5 and tumor
invasion. However, a correlation was indicated in rheumatoid
arthritis, an inflammatory disease. The PDCD5 levels in the plasma
and synovial fluid of patient with rheumatoid arthritis were shown
to be inversely associated with two inflammatory cytokines, tumor
necrosis factor (TNF)-α and interleukin (IL)-17 (23,24).
TNF-α-mediated nuclear factor-κB expression promotes the invasion
of HCC cells, and its downregulation mediates the anti-invasive
effect of a number of drugs (25).
IL-17 can also promote the invasion and metastasis of HCC cells
(26).
EMT is one vital step in epithelial cells,
characterized by loss of cell adhesion and acquisition of malignant
phenotype, including capabilities of cell motility, migration,
invasion and metastasis to a new location (27). The present results showed that
following TGF-β treatment, compared with the HepG2-Neo cells, the
HepG2-PDCD5 cells showed decreased expression levels of Snail,
which is an EMT marker protein. This means that PDCD5 has an
inhibitory effect on EMT in HCC cells, and provided a novel
anti-tumor strategy for treating HCC.
The present study found that TGF-β treatment
significantly increased IGF-1 protein expression in the HepG2-Neo
cells, which was attenuated in the HepG2-PDCD5 cells, indicating
negative regulation on IGF-1 by PDCD5. IGF-1 is mainly secreted by
the liver as a result of stimulation by growth hormone. IGF-1 plays
roles in the promotion of cell proliferation and the inhibition of
apoptosis. In human prostate cancer cells, IGF-1 upregulates ZEB1
and drives EMT (28). The present
results validated the effect of IGF-1 on EMT in HCC cells.
Recently, a study showed negative correlations between IGF-1 and
PDCD5 at the mRNA and protein levels (29). The study inferred that IGF-1 may
downregulate the expression of PDCD5. However, the present results
indicate that IGF-1 may be downregulated by PDCD5. This disparity
requires further investigation.
In conclusion, stable transfection of the PDCD5 gene
can inhibit the growth and induce cell cycle arrest in the
G2/M phase in HepG2 cells. PDCD5 transfection can also
enhance the sensitivity to cisplatin and p53 phosphorylation, and
reverse invasion and EMT induced by TGF-β. PDCD5 represents a novel
drug target and therapeutic strategy for the improved
chemotherapeutic treatment of HCC.
References
|
1
|
Khan N, Afaq F and Mukhtar H: Apoptosis by
dietary factors: the suicide solution for delaying cancer growth.
Carcinogenesis. 28:233–239. 2007. View Article : Google Scholar
|
|
2
|
Liekens S: Regulation of cancer
progression by inhibition of angiogenesis and induction of
apoptosis. Verh K Acad Geneeskd Belg. 70:175–191. 2008.PubMed/NCBI
|
|
3
|
Chen Y, Wang Y, Song H, Wang J, Yang H,
Xia Y, Xue J, Li S, Chen M and Lu Y: Expression profile of
apoptosis-related genes potentially explains early recurrence after
definitive chemoradiation in esophageal squamous cell carcinoma.
Tumour Biol. 35:4339–4346. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu H, Wang Y, Zhang Y, Song Q, Di C, Chen
G, Tang J and Ma D: TFAR19, a novel apoptosis-related gene cloned
from human leukemia cell line TF-1, could enhance apoptosis of some
tumor cells induced by growth factor withdrawal. Biochem Biophys
Res Commun. 254:203–210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Y, Sun R, Han W, Zhang Y, Song Q, Di
C and Ma D: Nuclear translocation of PDCD5 (TFAR19): an early
signal for apoptosis? FEBS Lett. 509:191–196. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Du YJ, Xiong L, Lou Y, Tan WL and Zheng
SB: Reduced expression of programmed cell death 5 protein in tissue
of human prostate cancer. Chin Med Sci J. 24:241–245. 2009.
View Article : Google Scholar
|
|
7
|
Spinola M, Meyer P, Kammerer S, Falvella
FS, Boettger MB, Hoyal CR, Pignatiello C, Fischer R, Roth RB,
Pastorino U, Haeussinger K, Nelson MR, Dierkesmann R, Dragani TA
and Braun A: Association of the PDCD5 locus with lung cancer risk
and prognosis in smokers. J Clin Oncol. 24:1672–1678. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang X, Wang X, Song X, Wei Z, Zhou C,
Zhu F, Wang Q, Ma C and Zhang L: Clinical and prognostic
significance of lost or decreased PDCD5 expression in human
epithelial ovarian carcinomas. Oncol Rep. 25:353–358. 2011.
View Article : Google Scholar
|
|
9
|
Li H, Wang Q, Gao F, Zhu F, Wang X, Zhou
C, Liu C, Chen Y, Ma C, Sun W and Zhang L: Reduced expression of
PDCD5 is associated with high-grade astrocytic gliomas. Oncol Rep.
20:573–579. 2008.PubMed/NCBI
|
|
10
|
Ruan GR, Qin YZ, Chen SS, Li JL, Ma X,
Chang Y, Wang YZ, Fu JY and Liu YR: Abnormal expression of the
programmed cell death 5 gene in acute and chronic myeloid leukemia.
Leuk Res. 30:1159–1165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yin A, Jiang Y, Zhang X, Zhao J and Luo H:
Transfection of PDCD5 sensitizes colorectal cancer cells to
cisplatin-induced apoptosis in vitro and in vivo. Eur J Pharmacol.
649:120–126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu HY, Chen ZW, Pan YM, Fan L, Guan J and
Lu YY: Transfection of PDCD5 effect on the biological behavior of
tumor cells and sensitized gastric cancer cells to
cisplatin-induced apoptosis. Dig Dis Sci. 57:1847–1856. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H, Zhang X, Song X, Zhu F, Wang Q, Guo
C, Liu C, Shi Y, Ma C, Wang X and Zhang L: PDCD5 promotes
cisplatin-induced apoptosis of glioma cells via activating
mitochondrial apoptotic pathway. Cancer Biol Ther. 13:822–830.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo XL, Ma NN, Zhou FG, Zhang L, Bu XX,
Sun K, Song JR, Li R, Zhang BH, Wu MC and Wei LX: Up-regulation of
hTERT expression by low-dose cisplatin contributes to chemotherapy
resistance in human hepatocellular cancer cells. Oncol Rep.
22:549–556. 2009.PubMed/NCBI
|
|
15
|
Asechi H, Hatano E, Nitta T, Tada M,
Iwaisako K, Tamaki N, Nagata H, Narita M, Yanagida A, Ikai I and
Uemoto S: Resistance to cisplatin-induced apoptosis via
PI3K-dependent survivin expression in a rat hepatoma cell line. Int
J Oncol. 37:89–96. 2010.PubMed/NCBI
|
|
16
|
Ando T, Kawabe T, Ohara H, Ducommun B,
Itoh M and Okamoto T: Involvement of the interaction between p21
and proliferating cell nuclear antigen for the maintenance of G2/M
arrest after DNA damage. J Biol Chem. 276:42971–42977. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Selvendiran K, Tong L, Vishwanath S,
Bratasz A, Trigg NJ, Kutala VK, Hideg K and Kuppusamy P: EF24
induces G2/M arrest and apoptosis in cisplatin-resistant human
ovarian cancer cells by increasing PTEN expression. J Biol Chem.
282:28609–28618. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weir NM, Selvendiran K, Kutala VK, Tong L,
Vishwanath S, Rajaram M, Tridandapani S, Anant S and Kuppusamy P:
Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant
human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer
Biol Ther. 6:178–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu XL, Xing BC, Han HB, Zhao W, Hu MH, Xu
ZL, Li JY, Xie Y, Gu J, Wang Y and Zhang ZQ: The properties of
tumor-initiating cells from a hepatocellular carcinoma patient’s
primary and recurrent tumor. Carcinogenesis. 31:167–174. 2010.
View Article : Google Scholar
|
|
20
|
Yao H, Feng Y, Zhou T, Wang J and Wang ZX:
NMR studies of the interaction between human programmed cell death
5 and human p53. Biochemistry. 51:2684–2693. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu L, Hu J, Zhao Y, Hu J, Xiao J, Wang Y,
Ma D and Chen Y: PDCD5 interacts with p53 and functions as a
positive regulator in the p53 pathway. Apoptosis. 17:1235–1245.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fransvea E, Mazzocca A, Antonaci S and
Giannelli G: Targeting transforming growth factor (TGF)-betaRI
inhibits activation of beta1 integrin and blocks vascular invasion
in hepatocellular carcinoma. Hepatology. 49:839–850. 2009.
View Article : Google Scholar
|
|
23
|
Wang J, Guan Z and Ge Z: Plasma and
synovial fluid programmed cell death 5 (PDCD5) levels are inversely
associated with TNF-α and disease activity in patients with
rheumatoid arthritis. Biomarkers. 18:155–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang JF, Guan ZP, Zhang SL, Pei Z, Chen YY
and Pan H: Programmed cell death 5 correlates with disease activity
and interleukin-17 in serum and synovial fluid of rheumatoid
arthritis patients. Chin Med J (Engl). 126:296–299. 2013.
|
|
25
|
Yu H, Pan C, Zhao S, Wang Z, Zhang H and
Wu W: Resveratrol inhibits tumor necrosis factor-alpha-mediated
matrix metalloproteinase-9 expression and invasion of human
hepatocellular carcinoma cells. Biomed Pharmacother. 62:366–372.
2008. View Article : Google Scholar
|
|
26
|
Li J, Lau GK, Chen L, Dong SS, Lan HY,
Huang XR, Li Y, Luk JM, Yuan YF and Guan XY: Interleukin 17A
promotes hepatocellular carcinoma metastasis via NF-κB induced
matrix metalloproteinases 2 and 9 expression. PLoS One.
6:e218162011. View Article : Google Scholar
|
|
27
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Graham TR, Zhau HE, Odero-Marah VA,
Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW and O’Regan
RM: Insulin-like growth factor-I-dependent up-regulation of ZEB1
drives epithelial-to-mesenchymal transition in human prostate
cancer cells. Cancer Res. 68:2479–2488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yi C, Ma C, Xie Z, Zhang G, Song W, Zhou X
and Cao Y: Down-regulation of programmed cell death 5 by
insulin-like growth factor 1 in osteoarthritis chondrocytes. Int
Orthop. 37:937–943. 2013. View Article : Google Scholar : PubMed/NCBI
|