Introduction
The death-associated protein kinase-1 (DAPK1)
gene is localized to chromosome 9q34.1 and encodes a 160-kDa
serine/threonine, microfilament-bound kinase which is involved in
interferon-γ, tumor necrosis factor-α and Fas ligand-induced
apoptosis, anoikis and autophagic cell death, respectively
(1–4). The DAPK1 protein has a kinase domain,
a calmodulin regulatory domain, ankirin repeats, P-loops, a
microfilament-binding domain and a death domain (5), allowing it to be involved in various
signaling pathways within the cell. For example, DAPK1 interacts
with extracellular signal-regulated kinase 1/2 (ERK1/2) via its
death domain. This interaction causes ERK to induce DAPK1
phosphorylation at Ser 735, which enhances the catalytic activity
of DAPK1. DAPK1 activity contributes to the arrest of ERK in the
cytoplasm, thus, blocking cell proliferation regulated by the
RAS/RAF/ERK signaling pathway. This reciprocal relationship between
DAPK1 and ERK may be involved in the regulation of apoptosis
(6).
DNA methylation occurs within CpG dinucleotides of
the mammalian genome. DNA methyltransferase enzymes (DNMTs)
catalyze the addition of a methyl group (−CH3) to the 5′
position of cytosine, resulting in methylated cytosine, termed
5-methylcytosine (5meC) (7). DNA
methylation is hypothesized to be involved in transcriptional
silencing (8), and loss of DNA
methylation appears to be associated with cellular differentiation
(9–11) and cancer growth (12–14).
Specific agents, such as 5-azacytidine and 5-aza-2′-deoxycytidine
(decitabine), inhibit DNA methylation by blocking DNMT activity
(15,16) and have been proposed for use in
cancer therapy (15,17–19).
DAPK1 methylation may be associated with the
loss of DAPK1 activity, as increased methylation in the
DAPK1 promoter region has been detected in various types of
cancer, such as renal (20) and
cervical cancers (21), B cell
lymphoma (22), myelodysplastic
syndrome, acute myeloblastic leukemia (23) and chronic myeloid leukemia (CML)
(24–26). CML is a myeloproliferative disorder
resulting from the oncogenic transformation of hematopoietic stem
cells, and is characterized by the Philadelphia chromosome, a
reciprocal translocation between the exon 2 sequence upstream of
the Abelson murine leukemia (ABL) proto-oncogene on
chromosome 9 and the 5′ sequence of the breakpoint cluster region
(BCR) gene on chromosome 22. The transcript of oncogenic
BCR-ABL is a 210-kDa protein with tyrosine kinase activity
that is present in the cytoplasm and activates mitogenic and
anti-apoptotic pathways (27,28).
Imatinib (STI571, Gleevec®, Glivec®) is
typically used to inhibit the tyrosine kinase activity of the
BCR-ABL protein in CML therapy; however, specific CML patients are
unresponsive to imatinib treatment (29). Mutations within BCR-ABL cause
increased BCR-ABL expression levels and, therefore, these patients
consequently develop imatinib resistance. These mutations include
T315I (in the imatinib-binding domain of BCR-ABL), M351T (in the
catalytic domain) and E255K (in the ATP-binding domain) (30,31).
The present study aimed to investigate whether
DAPK1 methylation occurs in CML patients with or without
imatinib resistance, and identified that: i) The DAPK1
promoter was significantly methylated in CML patients (10/43)
compared with healthy individuals (0/25); ii) the proportion of
imatinib-resistant CML patients demonstrating DAPK1
methylation (6/17) was higher than the proportion of non-resistant
CML patients demonstrating DAPK1 methylation (4/26); and
iii) the incidence of DAPK1 methylation in resistant
patients varied between the different types of BCR-ABL
mutation. The results of the present study indicate that
DAPK1 methylation may be associated with resistance to
imatinib therapy in CML patients; however, this is dependent on the
type of mutation causing the resistance.
Materials and methods
Samples
Blood samples were obtained from 43 CML adults who
had enrolled in clinical assessment for imatinib therapy. The
samples were screened for resistance to imatinib and for the
presence of DAPK1 methylation. Additionally, control blood
samples were collected from 25 healthy adults. All participants
were enrolled at the Department of Medical Biology, Faculty of
Medicine, Ankara University (Ankara, Turkey). Informed consent was
obtained from all participants and the research protocol was
approved by Ankara No. 1 Clinical Research Ethics Committee
(Ankara, Turkey).
DNA isolation
DNA samples from peripheral blood were isolated
using the salt precipitation method. Briefly, the cells were lysed
on ice for 1 h in 1.54 M lysis buffer, followed by
incubation with 1X sodium chloride-tris-EDTA and 10% SDS (Fisher
Scientific, Pittsburgh, PA, USA), and incubated with 0.865 M
proteinase K (Sigma, St. Louis, MO, USA) at 37°C overnight. Whole
blood cells were subsequently treated with 5.6 M NaCl and
centrifuged at 750 × g for 20 min, and the resultant DNA samples
were incubated overnight in distilled water at 37°C.
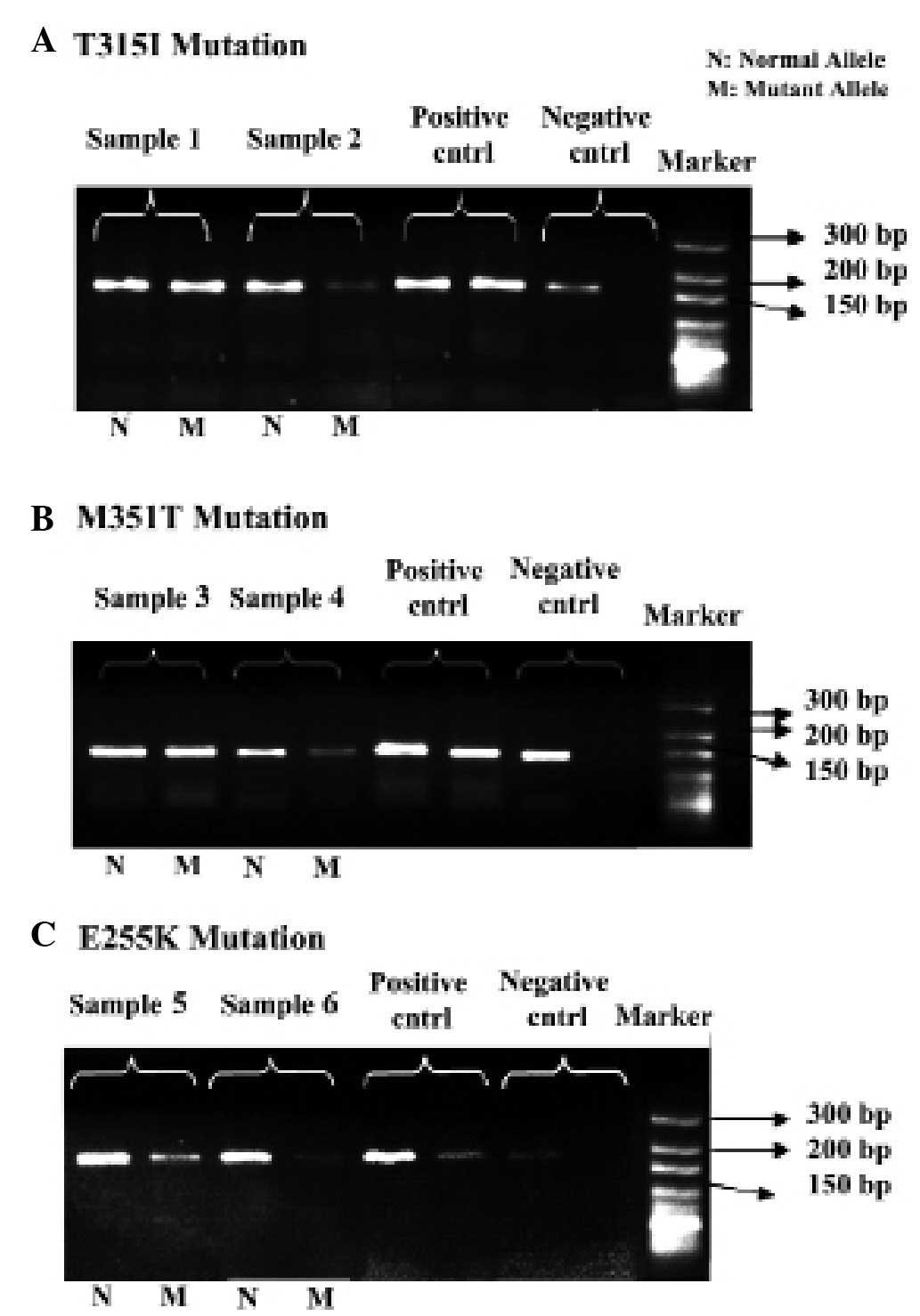
BCR-ABL mutation analysis
Mutations conferring imatinib resistance were
detected using allele-specific oligonucleotide polymerase chain
reaction (ASO-PCR). PCR reactions for T315I, M351T (33) and E255K (33) were performed as previously
described. The primer sequences for T315I were as follows: Forward,
5′-GCC CCC CTT CTA TAT CAT CAC-3′ for normal PCR; forward, 5′-GCC
CCC CTT CTA TAT CAT CAT-3′ for ASO-PCR; and reverse, 5′-GGA TGA AGT
TTT TCT TCT CCA-3′. The primer sequences for M351T were as follows:
Forward, 5′-CCA CTC AGA TCT CGT CAG CCA T-3′ for normal PCR;
forward, 5′-CCA CTC AGA TCT CGT CAG CCA C-3′ for ASO-PCR; and
reverse, 5′-GCC CTG AGA CCT CCT AGG CT-3′. The primer sequences for
E255K were as follows: Forward, 5′-GCG GGG GCC AGT ACG GGG-3′ for
normal PCR; forward, 5′-GCG GGG GCC AGT ACG GGA-3′ for ASO-PCR; and
reverse, 5′-GCC AAT GAA GCC CTC GGA C-3′. The predicted PCR
products are 158, 149 and 192 bp for T315I, M351T and E255K,
respectively.
Sodium bisulfite modification of DNA
A CpGenome™ DNA Modification kit (cat. no. S7820;
EMD Millipore, Billerica, MA, USA) was used to modify the DNA,
according to the manufacturer’s instructions. Briefly, this
included reagent preparation, DNA modification, initial desalting,
the completion of DNA modification (desulfonation), second
desalting and elution.
Methylation-specific PCR (MS-PCR)
A CpG WIZ® DAP-Kinase Amplification kit
(cat. no. S7801; EMD Millipore) was used to amplify the DNA. The
primer sequences designed for the DAPK1 promoter are
indicated in Fig. 1Aa. The
amplification kit included unmethylated (U), methylated (M) and
wild-type (W) DNA. Prior to performing PCR, the U and M DNAs were
sodium bisulfite-modified. MS-PCR was conducted by performing 40
cycles of 95°C for 5 min, 95°C for 45 sec, 58°C for 45 sec and 72°C
for 60 sec, and the PCR products for the M and U alleles were
finally defined by performing 2% agarose gel electrophoresis. The
predicted PCR products are 105, 97 and 99 bp for U, M and W DNA,
respectively.
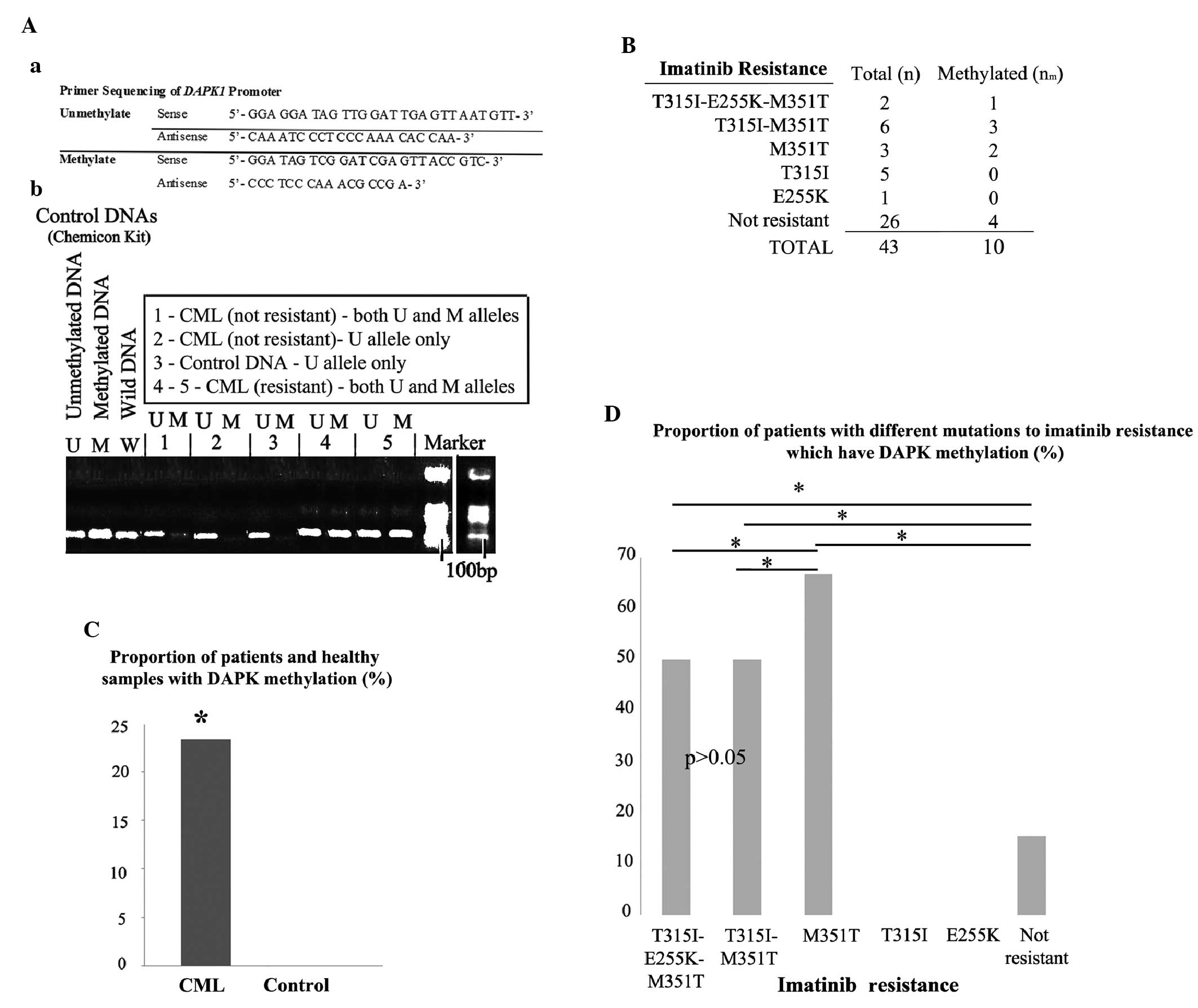 | Figure 1Analysis of DAPK1 methylation.
(Aa) DAPK primer sequences used for methylation analysis
performed using methylation-specific PCR following bisulfite
conversion. (Ab) Representative PCR products for U and M alleles of
DAPK1 in resistant (samples 4 and 5) and non-resistant
(samples 1 and 2) patients, in a control sample from healthy
individuals (sample 3), as well as in control U, M and W DNA
samples. (B) The total number of samples and the number of samples
exhibiting DAPK1 methylation. (C) The proportion (%) of
samples with DAPK1 methylation is significantly greater in
CML patients compared with the healthy controls. (D) The proportion
(%) of patients with DAPK1 methylation varied among different
mutations. The majority of patients with M351T demonstrated
DAPK1 methylation; however, none of patients with T315I or
E255K were methylated, and no difference was detected between the
patients with triple (T315I-M351T-E255K) and double (T315I-M351T)
mutations (P>0.05). Furthermore, a relatively low number of
non-resistant patients demonstrated DAPK1 methylation
(P<0.05). *P<0.05. DAPK1, death-associated
protein kinase-1; PCR, polymerase chain reaction; U, unmethylated;
M, methylated; W, wild type; CML, chronic myeloid leukemia. |
Statistical analysis
Statistical analysis was performed using SPSS
software (version 17.0; SPSS, Inc., Chicago, IL, USA) and graphs
were constructed using SPSS or Microsoft Excel software (Microsoft
Corporation, Redmond, WA, USA). The proportion (%) of samples
demonstrating DAPK1 methylation was arcsine-transformed and
compared using a Mann-Whitney U test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Determination of imatinib resistance
mutations
Initially, the mutation profiles for the
imatinib-resistant CML patients (determining the existence of
T315I, M351T and E255K mutations) were examined using ASO-PCR. The
cohort was assembled from numerous CML patients who had experienced
failed therapies at various stages of the disease and, therefore,
had applied to The Department of Medical Biology, Faculty of
Medicine, Ankara University (Ankara, Turkey) for investigation into
imatinib unresponsiveness. As predicted, these patients were
determined to have at least one of the resistance mutations
examined and, as such, were described as resistant to imatinib
(Fig. 2A–C). The patients who were
clinically responsive to imatinib were determined to have normal
BCR-ABL alleles and were described as non-resistant to
imatinib. Imatinib resistance was detected regardless of the stage
of CML and, therefore, patients were divided into just two groups:
Resistant and non-resistant.
Methylation analysis of the DAPK1
gene
Subsequently, the existence of methylation in the
promoter region of the DAPK1 gene was determined using
sodium bisulfite modification of the DNA samples, followed by
MS-PCR. The primer sequences designed for the U and M alleles of
the DAPK1 promoter region are demonstrated in Fig. 1Aa, and representative PCR products
for the U and M alleles of the DAPK1 gene are demonstrated
in Fig. 1Ab. The patients with a U
allele alone were described as not methylated (i.e., samples 2 and
3) and the patients exhibiting an additional M allele (i.e.,
samples 1, 4 and 5; Fig. 1Ab) were
described as methylated. As expected, none of the healthy
individuals (control; n=0/25) exhibited a methylated DAPK1
promoter region; however, almost 25% of CML patients (resistant and
non-resistant) exhibited DAPK1 methylation (P<0.05;
Fig. 1B). The proportion of
patients with or without DAPK1 methylation are provided in
Fig. 1C. No methylation was
detected in patients with T315I (0/5) or E255K (0/1), and 4/26
non-resistant patients demonstrated DAPK1 methylation
(Fig. 1C). A detailed comparison of
the proportion (%) of patients with DAPK1 methylation
between the different BCR-ABL mutation groups is indicated in
Fig. 1D. Compared with the other
mutation groups, the highest proportion of DAPK1 methylation
was detected in the M351T alone mutation group (P<0.05) and the
lowest proportion was observed in the non-resistant patients
(P<0.05; Fig. 1D). However, no
significant difference was identified in the proportion (%) of
patients with DAPK1 methylation between those with triple
(T315I, E255K and M351T) and double (T315I and M351T) mutations
(P>0.05; Fig. 1D). Furthermore,
no methylation was detected in patients exhibiting T315I or E255K
alone (Fig. 1D).
Discussion
The DAPK1 protein is known to be involved in the
suppression of cancer formation and metastasis via apoptosis and,
thus, is considered to be a tumor suppressor gene (1). CpG methylation in the DAPK1
promoter region has been detected in a range of solid cancers, such
as non-small cell lung cancer (34), leiomyosarcoma (35), nasopharyngeal carcinoma (36), and hematological malignancies, such
as follicular lymphoma (37) and
CML (24,25,38). A
previous study determined that 50% of CML patients exhibited
DAPK1 methylation, and this was not correlated with age,
hematological parameters, chromosomal abnormalities or the type and
quantity of the BCR/ABL transcripts, however, it was
correlated with gender and CML phase (26). In intestinal system cancers,
DAPK1 activity was inhibited by a protein complex that
included DNMT1. Furthermore, deacetylation of histones H3 and H4
appeared to contribute to DAPK1 silencing (39). These factors indicate that the
silencing of DAPK1 by such methylation may be associated
with cancer progression. To the best of our knowledge, however, no
study has been conducted investigating the correlation between the
presence of methylation in the DAPK1 promoter region and
mutations that confer resistance to imatinib therapy in CML
patients. The present study examined whether DAPK1
methylation occurred in CML patients with or without resistance to
imatinib. DNA methylation (resulting in 5meC) is typically detected
using bisulfite sequencing. This methodology is based on the
discrimination between methylated and unmethylated cytosines by
treatment with sodium bisulfite followed by MS-PCR. However, with
the discovery of novel 5meC modifications [for example, 5hmC
(5-hydroxymethylcytosine)] (40–42),
it has been demonstrated that standard sodium bisulfite treatment
is unable to distinguish between 5meC and 5hmC (43,44).
Following bisulfite treatment, 5hmC is converted to
cytosine-5-methylensulfonate (CMS) and CMS is read as 5meC
(43). The development of an
additional DNA treatment is therefore required for the accurate
discrimination of 5meC from other DNA modifications. Various
attempts to improve this discrimination with additional steps have
been conducted, for example, ten-eleven translocation
methylcytosine dioxygenase (TET)-assisted bisulfite sequencing,
which includes glucosylation and TET oxidation of genomic DNA,
resulting in the discrimination of 5meC from 5hmC (45,46);
the conversion of 5hmC to CMS to provide genome scale profile of
5hmC (47); and chemical
modification of 5-carboxylcytosine (5caC) to provide base
resolution detection of 5caC (48).
Gold standard bisulfite-based methodologies using a standard sodium
bisulfite treatment are able to reveal an overall profile of 5meC
modifications, rather than a 5meC profile alone, although caution
is required when interpreting the changes in DNA methylation as
they do not reflect the changes in the individual 5meC metabolites.
The present study identified that the DAPK1 gene is
significantly methylated (including possible hydroxymethylation) in
CML patients and this is correlated with mutations that result in
resistance to imatinib. Notably, one in two of the patients
exhibiting both T315I and M351T mutations demonstrated DNA
methylation in the DAPK1 promoter region compared with
non-resistant patients; however, none of the patients with T315I
alone demonstrated DAPK1 methylation. Patients exhibiting
the E255K mutation alone or with other mutations were not
identified to have DAPK1 methylation. Furthermore, it was
observed that the PCR product bands of the M alleles for the
DAPK1 promoter were thicker in the resistant CML patients
compared with the non-resistant patients (and a similar variation
was observed in the PCR products of the various resistance
mutations). Although the band thickness may indicate the difference
in the level of methylation of the DAPK1 gene, the present
study did not explore the extent of methylation but instead aimed
to identify whether methylation exists in the DAPK1 promoter
region. The results of the present study may indicate that
methylation of the DAPK1 promoter region is associated with
a specific signaling pathway(s) in the resistance to imatinib.
Imatinib resistance can be induced by point
mutations (such as T315I and M351T) of transgenic BCR-ABL
and this results in increased expression of the oncogenic BCR-ABL
fusion protein. BCR-ABL may be involved in various cell
proliferation pathways within the cells, such as rat sarcoma (RAS),
Janus kinase/signal transducer and activator of transcription,
phosphatidylinositol 3-kinase and Myc (1,49). The
protein complex of BCR-ABL with growth factor receptor bound
protein 2 and son of sevenless activates the RAS pathway (49), and RAS activates mitogen-activated
protein kinase (MAPK) and MEK1/2 (a MAPK kinase), resulting in the
transportation of extracellular signal-regulated kinase (ERK) from
the cytosol to the nucleus (1).
Furthermore, DAPK1 induces apoptosis when it is phosphorylated by
the RAS-ERK signaling pathway (50). The death domain of the DAPK1 protein
interacts with ERK (6) and ERK is
arrested in the cytoplasm. Thus, the increase in the expression
level of the BCR-ABL transcript may not be eliminated if the
DAPK1-regulated apoptosis pathway is inhibited. The results
of the present study indicate that DAPK1 methylation may be
involved in imatinib resistance in CML depending on the type of
BCR-ABL mutation; however, DAPK1 may not have a direct
effect on the suppression of overexpressed tyrosine kinase in CML
patients. Furthermore, it is possible that imatinib resistance may
not depend on BCR-ABL activity (51); therefore, the proposed contribution
of DAPK1 to the BCR-ABL pathway requires a detailed investigation.
An additional study may be required to investigate whether imatinib
resistance induces DAPK1 methylation or vice versa.
Regarding the possible involvement of DNA
methylation in CML, the additional use of demethylating
agents (such as decitabine) may be useful in CML treatment. The
level of global methylation (as assessed by the methylation of the
retrotransposable element of the human genome, long interspersed
element-1 gene) was relatively greater in patients responding to a
combined treatment of imatinib with decitabine compared with
non-responder patients (18,19);
furthermore, patients without imatinib resistance demonstrated a
higher rate of response to this combination therapy (19). This indicates that patients with
imatinib resistance may be less responsive to the combined use of
decitabine with imatinib. However, the additional use of a
demethylating agent with imatinib may induce apoptosis in CML, as
this combined treatment inhibited imatinib resistant cell growth
in vitro to some extent compared with decitabine treatment
alone (52). Therefore, the
detection of the methylation status of tumor suppressor genes (such
as those involved in apoptosis) may be important for the selection
of additional treatment with imatinib in CML therapy. The current
problem with the use of demethylating agents is that they induce
global rather than gene-specific demethylation, potentially
resulting in proto-oncogene activation by demethylation (53). Demethylation of the DAPK1
gene can be induced by a demethylating agent, allowing
imatinib-resistance to be overcome. This may be useful for
identifying whether DAPK1 demethylation induced by such
demethylating agents reduces the incidence of BCR-ABL mutation.
CML patients with T315I and E255K mutations were
detected to be insensitive to clinically achievable doses of
imatinib (54), indicating that
additional agents may be required for the treatment of these
patients. However, higher doses of imatinib were useful in cases of
M351T and Y253F mutation (54).
Therefore, the strategy for individual or combined treatment should
depend on the type of mutations causing resistance. The findings of
the present study indicate that the E255K mutation is not involved
in the association between imatinib resistance and DAPK1
methylation, instead, M351T is the major mutation in this
association. M351T (which occurs in the catalytic domain of
BCR-ABL) may be more emphasized than mutations in other domains as
it increases the catalytic function of kinases. However, it should
be noted that some other mutations in BCR-ABL that were not
examined in the present study (such as Y253H, F317L and H396R)
(31) require investigation to
achieve a broader comparison.
In conclusion, the current study presents a typical
trend for the association between the presence of DAPK1
promoter methylation and a variety of BCR-ABL mutations,
regardless of any characteristics of the CML patients, such tumor
stage, gender or age. The present study provides an insight into
the understanding of imatinib resistance in CML progression and
proposes that the determination of DAPK1 methylation may be
a criterion to use an additional agent in the treatment of CML
(i.e., decitabine). Furthermore, the methylation status of various
other tumor suppressor genes may be useful for the determination of
an accurate CML treatment strategy. This is important as cancer is
a complex disease formed by the genetic and epigenetic mechanisms
of multiple genes.
Acknowledgements
The authors of the present study thank Ibn-i Sina
Hospital of Ankara University for providing the blood samples, and
Dr Mehmet Taspinar and biologist Ms. Sibel Arat for collecting the
samples. The authors would also like to thank Dr Sevil Oskay
Halacli (Hacettepe University, Sihhiye, Turkey) for her
support.
References
|
1
|
Gozuacik D and Kimchi A: DAPk protein
family and cancer. Autophagy. 2:74–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hervouet E, Cheray M, Vallette F and
Cartron P: DNA methylation and apoptosis resistance in cancer
cells. Cells. 2:545–573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen RH, Wang WJ and Kuo JC: The tumor
suppressor DAP-kinase links cell adhesion and cytoskeleton
reorganization to cell death regulation. J Biomed Sci. 13:193–199.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tang X, Wu W, Sun SY, et al:
Hypermethylation of the death-associated protein kinase promoter
attenuates the sensitivity to TRAIL-induced apoptosis in human
non-small cell lung cancer cells. Mol Cancer Res. 2:685–691.
2004.
|
|
5
|
Raveh T and Kimchi A: DAP kinase - a
proapoptotic gene that functions as a tumor suppressor. Exp Cell
Res. 264:185–192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen C, Wang W, Kuo J, et al:
Bidirectional signals tranduced by DAPK-ERK interaction promote the
apoptotic effect of DAPK. EMBO J. 24:294–304. 2005. View Article : Google Scholar :
|
|
7
|
Grønbæk K, Hother C and Jones P:
Epigenetic changes in cancer. APMIS. 115:1039–1059. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raynal NJ, Si J, Taby RF, et al: DNA
methylation does not stably lock gene expression but instead serves
as a molecular mark for gene silencing memory. Cancer Res.
72:1170–1181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tarfiei G, Noruzinia M, Soleimani M, et
al: ROR2 promoter methylation change in osteoblastic
differentiation of mesenchymal stem cells. Cell J. 13:11–15.
2011.PubMed/NCBI
|
|
10
|
Nazor KL, Altun G, Lynch C, et al:
Recurrent variations in DNA methylation in human pluripotent stem
cells and their differentiated derivatives. Cell Stem Cell.
10:620–634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bocker MT, Hellwig I, Breiling A, et al:
Genome-wide promoter DNA methylation dynamics of human
hematopoietic progenitor cells during differentiation and aging.
Blood. 117:e182–e189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pattani K, Soudry E, Glazer C, et al:
MAGEB2 is activated by promoter demethylation in head and neck
squamous cell carcinoma. PLoS One. 7:e455342012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stefanska B, Huang J, Bhattacharyya B, et
al: Definition of the landscape of promoter DNA hypomethylation in
liver cancer. Cancer Res. 71:5891–5903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song IS, Ha GH, Kim JM, et al: Human
ZNF312b oncogene is regulated by Sp1 binding to its promoter region
through DNA demethylation and histone acetylation in gastric
cancer. Int J Cancer. 129:2124–2133. 2011. View Article : Google Scholar
|
|
15
|
Lavelle D, DeSimone J, Hankewych M,
Kousnetzova T and Chen YH: Decitabine induces cell cycle arrest at
the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP
kinase pathway. Leuk Res. 27:999–1007. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mohana Kumar B, Jin HF, Kim JG, et al: DNA
methylation levels in porcine fetal fibroblasts induced by an
inhibitor of methylation, 5-azacytidine. Cell Tissue Res.
325:445–454. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al-Romaih K, Sadikovic B, Yoshimoto M, et
al: Decitabine-induced demethylation of 5′ CpG island in GADD45A
leads to apoptosis in osteosarcoma cells. Neoplasia. 10:471–480.
2008.PubMed/NCBI
|
|
18
|
Issa J, Gharibyan V, Cories J, et al:
Phase II study of low-dose decitabine in patients with chronic
myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol.
23:3948–3956. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oki Y, Kantarjian H, Gharibyan V, et al:
Phase II study of low-dose decitabine in combination with imatinib
mesylate in patients with accelerated or myeloid blastic phase of
chronic myelogenous leukemia. Cancer. 109:899–906. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahmad S, Arjumand W, Seth A, Saini A and
Sultana S: Methylation of the APAF-1 and DAPK-1 promoter region
correlates with progression of renal cell carcinoma in North Indian
population. Tumor Biol. 33:395–402. 2012. View Article : Google Scholar
|
|
21
|
Banzai C, Nishnio K, Quan J, et al:
Gynecological Cancer Registry of Niigata: Promoter methylation of
DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. Int J
Clin Oncol. 19:127–132. 2014. View Article : Google Scholar
|
|
22
|
Kristensen LS, Treppendahl MB, Asmar F, et
al: Investigation of MGMT and DAPK1 methylation patterns in diffuse
large B-cell lymphoma using allelic MSP-pyrosequencing. Sci Rep.
3:27892013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Claus R, Hackanson B, Poetsch AR, et al:
Quantitative analyses of DAPK1 methylation in AML and MDS. Int J
Cancer. 131:E138–E142. 2012. View Article : Google Scholar :
|
|
24
|
Katzenellenbogen R, Baylin S and Herman J:
Hypermethylation of the DAP-kinase CpG island is a common
alteration in B-cell malignancies. Blood. 93:4347–4353.
1999.PubMed/NCBI
|
|
25
|
Mir R, Ahmad I, Javid J, et al: Epigenetic
silencing of DAPK1 gene is associated with faster disease
progression in India populations with chronic myeloid leukemia. J
Cancer Sci Ther. 5:144–149. 2013. View Article : Google Scholar
|
|
26
|
Qian J, Wang YL, Lin J, et al: Aberrant
methylation of the death-associated protein kinase 1 (DAPK1) CpG
island in chronic myeloid leukemia. Eur J Haematol. 82:119–123.
2009. View Article : Google Scholar
|
|
27
|
Yanagisawa K, Yamauchi H, Kaneko M, et al:
Suppression of cell proliferation and the expression of a bcr-abl
fusion gene and apoptotic cell death in a new human chronic
myelogenous leukaemia cell line, KT-1, by interferon-α. Blood.
91:641–648. 1998.PubMed/NCBI
|
|
28
|
Vigneri P and Wang JY: Induction of
apoptosis in chronic myelogenous leukaemia cells through nuclear
entrapment of BCR-ABL tyrosine kinase. Nat Med. 7:228–234. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Litzow MR: Imatinib resistance: obstacles
and opportunities. Arch Pathol Lab Med. 130:669–679.
2006.PubMed/NCBI
|
|
30
|
Walz C and Sattler M: Novel targeted
therapies to overcome imatinib mesylate resistance in chronic
myeloid leukaemia (CML). Crit Rev Oncol Hematol. 57:145–164. 2006.
View Article : Google Scholar
|
|
31
|
Corbin AS, Rosée P, Stoffregen EP, Druker
BJ and Deininger MW: Several Bcr-Abl kinase domain mutants
associated with imatinib mesylate resistance remain sensitive to
imatinib. Blood. 101:4611–4614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roche-Lestienne C, Soenen-Cornu V,
Grardel-Duflos N, et al: Several types of mutations of the Abl gene
can be found in chronic myeloid leukemia patients resistant to
STI571, and they can pre-exist to the onset of treatment. Blood.
100:1014–1018. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kang HY, Hwang JY, Kim SH, et al:
Comparison of allele specific oligonucleotide-polymerase chain
reaction and direct sequencing for high throughput screening of ABL
kinase domain mutations in chronic myeloid leukemia resistant to
imatinib. Haematologica. 91:659–662. 2006.PubMed/NCBI
|
|
34
|
Tang X, Khuri FR, Lee JJ, et al:
Hypermethylation of the death-associated protein (DAP) kinase
promoter and aggressiveness in stage non-small-cell lung cancer. J
Natl Cancer Inst. 92:1511–1516. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kawaguchi K, Oda Y, Saito T, et al:
Death-associated protein kinase (DAP kinase) alteration in soft
tissue leiomyosarcoma: Promoter methylation or homozygous deletion
is associated with a loss of DAP kinase expression. Hum Pathol.
35:1266–1271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wong TS, Chang HW, Tang KC, et al: High
frequency of promoter hypermethylation of the death-associated
protein-kinase gene in nasopharyngeal carcinoma and its detection
in the peripheral blood of patients. Clin Cancer Res. 8:433–437.
2002.PubMed/NCBI
|
|
37
|
Voso MT, Gumiero D, D’alo’ F, et al:
DAP-kinase hypermethylation in the bone marrow of patients with
follicular lymphoma. Haematologica. 91:1252–1256. 2006.PubMed/NCBI
|
|
38
|
Uehara E, Takeuchi S, Yang Y, et al:
Aberrant methylation in promoter-associated CpG islands of multiple
genes in chronic myelogenous leukemia blast crisis. Oncol Lett.
3:190–192. 2012.PubMed/NCBI
|
|
39
|
Satoh A, Toyota M, Itoh F, et al: DNA
methylation and histone deacetylation associated with silencing DAP
kinase gene expression in colorectal and gastric cancers. Br J
Cancer. 86:1817–1823. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He YF, Li BZ, Li Z, et al: Tet-Mediated
ormation of 5-carboxylcytosine and its excision by TDG in mammalian
DNA. Science. 333:1303–1307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ito S, Shen L, Dai Q, et al: Tet proteins
can convert 5-methylcytosine to 5-formylcytosine and
5-carboxylcytosine. Science. 333:1300–1303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tahiliani M, Koh KP, Shen YH, et al:
Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang Y, Pastor WA, Shen Y, et al: The
behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS
One. 5:e88882010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nestor C, Ruzov A, Meehan R and Dunican D:
Enzymatic approaches and bisulfite sequencing cannot distinguish
between 5-methylcytosine and 5-hydroxymethylcytosine in DNA.
Biotechniques. 48:317–319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yu M, Hon GC, Szulwach KE, et al:
Base-resolution analysis of 5-hydroxymethylcytosine in the
mammalian genome. Cell. 149:1368–1380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu M, Hon GC, Szulwach KE, et al:
Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat
Protoc. 7:2159–2170. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang Y, Pastor WA, Zepeda-Martínez JA and
Rao A: The anti-CMS technique for genome-wide mapping of
5-hydroxymethylcytosine. Nat Protoc. 7:1897–1908. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu X, Song CX, Szulwach K, et al: Chemical
modification-assisted bisulfite sequencing (CAB-Seq) for
5-carboxylcytosine detection in DNA. J Am Chem Soc. 135:9315–9317.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cilloni D and Saglio G: Molecular
pathways: BCR-ABL. Clin Cancer Res. 18:930–937. 2012. View Article : Google Scholar
|
|
50
|
Anjum R, Roux PP, Ballif BA, Gygi SP and
Blenis J: The tumor suppressor DAP kinase is a target of
RSK-mediated survival signaling. Curr Biol. 15:1762–1767. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Quentmeier H, Eberth S, Romani J, Zaborski
M and Drexler HG: BCR-ABL1-independent PI3Kinase activation causing
imatinib-resistance. J Hematol Oncol. 4:62011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
La Rosée P, Johnson K, Corbin AS, et al:
In vitro efficacy of combined treatment depends on the underlying
mechanism of resistance in imatinib-resistant Bcr-Abl-positive cell
lines. Blood. 103:208–215. 2004. View Article : Google Scholar
|
|
53
|
Howell PM Jr, Liu Z and Khong HT:
Demethylating agents in the treatment of cancer. Pharmaceuticals.
3:2022–2044. 2010. View Article : Google Scholar
|
|
54
|
Hochhaus A, Kreil S, Corbin AS, et al:
Molecular and chromosomal mechanisms of resistance to imatinib
(STI571) therapy. Leukemia. 16:2190–2196. 2002. View Article : Google Scholar : PubMed/NCBI
|