Introduction
Lentiviral vectors (LvVs) have emerged as an
important tool for transgene delivery and gene therapy. LvVs offer
various advantages for gene therapy due to their ability to infect
quiescent, slowly dividing or non-dividing cells, such as
hematopoietic stem cells, neurons and glial cells; their ability to
integrate into the host cell genome, resulting in long-term
transgene expression; and their large packaging capacity (1). Integrated LvVs do not induce an
inflammatory or immune response, which allows long-term in
vivo maintenance of transgene expression in a variety of tissue
types.
Standard LvV production typically relies on the
transient transfection of human embryonic kidney cells (HEK 293T)
with a packaging plasmid, an envelope glycoprotein-encoding plasmid
and a lentiviral transfer vector plasmid (2). Following transfection, lentiviral
particles (LvPs) are produced and released into the culture
supernatant of the HEK 293T cells. Although novel methods of
producing of LvVs have been developed (3), and various transfection protocols
using specific transfection reagents, such as
Lipofectamine® 2000 (4),
SuperFect® (5),
FuGENE® 6 and GeneJammer (6), have been successfully applied to the
production of LvVs, high costs have hindered their extensive use.
Thus, the transient transfection method using CaPO4
precipitation remains the most common method used for the
production of LvVs (7). The main
limitations of the CaPO4-mediated method of transient
transfection are the large quantities of DNA required and the
dependency of the method on an optimal pH of HEPES buffer. This pH
can drift over time, resulting in large variability in transfection
efficiency and significant batch-to-batch variation of the LvV
titers (8). Furthermore, despite
their safety and the marked progress that has been made in the
development of lentiviral packaging systems, the average range of
titers of crude lentiviral vector stocks produced is between
106 and 108 transduction units (TU) per ml
(9–10). This range is useful for in
vitro applications; however, the quantities are not high enough
for in vivo use. For in vivo applications, large
volumes of medium, large numbers of cells and large amounts of
plasmid DNA are required to prepare sufficient stocks of crude
LvVs. In addition, the relatively low transduction efficiency of
LvVs means that the production of high-titer, high-quality LvV
stocks is difficult and costly (11). Thus, improved methods for the
consistent production of high-titer LvVs are required for the
feasible use of LvVs in transgene delivery and gene therapy.
Previously, the polyethylenimine (PEI)-mediated transient
transfection method for LvV production was described (12); it uses a transfection reagent to
produce recombinant viral vectors, such as adeno-associated viral
vectors (AVVs) (13–14) or lentiviral vectors (12,15).
There are various advantages of producing LvVs via the PEI-mediated
transfection method. First, the relatively low cost of PEI allows
it to be widely used in the research laboratory. Second, the
chemical stability and pH independence of PEI ensures consistent
high transfection efficiency (16–17).
Finally, PEI has been demonstrated to be effective in transducing
adherent and suspension cells (18–19).
The aim of the present study was to optimize the
PEI-mediated transfection method in order to simplify the efficient
production of LvVs, and to compare the CaPO4- and
PEI-mediated transfection methods for producing LvVs. The
PEI-mediated method was identified to be simple to use and more
reliable, with a high degree of reproducibility and consistency.
Despite using less DNA, the PEI-mediated transfection method
resulted in viral titers that were the same as those achieved using
the CaPO4-mediated method.
Materials and methods
Experimental cell lines
HEK 293T cells and murine fibroblastic NIH 3T3 cells
were stocked in Dr Barbara C. Vanderhyden’s laboratory at the
University of Ottawa (Ottawa, Ontario, Canada)and cultured in
HyClone™ Dulbecco’s modified Eagle medium (DMEM)/high glucose
(Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; GE Healthcare Bio-Sciences, Pittsburgh,
PA, USA). The cells were maintained at 37°C and 5% CO2
in a humidified incubator (VWR International, LLC., Atlanta, GA,
USA) and were harvested at 80–90% confluence.
Plasmid constructs
The present study used a lentiviral transfer vector
plasmid (pWPI) containing enhanced green fluorescent protein
(eGFP), a pCMV-dR8.74 packaging plasmid and a pCAG4-Eco envelope
plasmid, as previously described (20–21).
pWPI and pCMV-dR8.74 plasmids were provided by Dr. D Trono (Swiss
Federal Institute of Technology in Lausanne, Lausanne, Switzerland)
and pCAG4-Eco plasmids were provided by Dr. A Nienhuis (St. Jude
Children’s Research Hospital, Memphis, TN, USA).
Preparation of PEI stocks
A 7.5 mM PEI stock solution was prepared by
dissolving 0.15 g PEI (molecular weight, 40 kDa; Polysciences Inc.,
Warrington, PA, USA) in 245 ml tissue culture grade water. Once
prepared, the PEI stock was stored at 4°C (22).
PEI-mediated transfections were performed
to generate lentiviral vectors
Lentiviral vectors were generated by the transient
cotransfection of HEK 293T cells with a three-plasmid system. The
lentiviral transfer vector plasmid (pWPI; 2 μg), packaging plasmid
(pCMV-dR8.74; 1.3 μg) and envelope plasmid (pCAG4-Eco; 1.25 μg) DNA
were mixed in 150 mM NaCl. Concurrently, 7.5 mM PEI stock was
diluted 6-fold with 150 mM NaCl to yield a final PEI concentration
of 1.25mM, using the formula previously described by Reed et
al (13), and 0.38 ml was added
to the 0.38 ml DNA (4.55 μg total) from above. Transfections were
also performed using 2× DNA (9.10 μg) and 3× DNA (13.65) solutions.
DNA/PEI mixtures were incubated at room temperature for ≥10 min.
The HEK 293T cells were trypsinized, washed twice with 1X phosphate
buffered saline (PBS) and resuspended (2×106 cells/ml)
in serum-free Opti-MEM® (Invitrogen Life Technologies,
Inc., Carlsbad, CA, USA). The DNA/PEI mixture was added to 7.5 ml
suspended cells, and immediately plated onto a 10 cm and incubated
at 37°C in a 5% CO2 humidified atmosphere. The
supernatants containing LvVs were collected and stored at
−80°C.
CaPO4-mediated transfections
were performed to generate LvVs
HEK 293T cells (2×106 cells/well) were
seeded in each 10 cm culture dish and incubated at 37°C in a 5%
CO2 humidified atmosphere. After 24 h, transfer vector
plasmid (pWPI; 20 μg), packaging plasmid (pCMV-dR8.74; 15 μg) and
envelope plasmid (pCAG4-Eco; 6 μg) were diluted with 250 μl sterile
double distilled water (Barnstead™ Nanopure™ ultrapure water
purification system; Thermo Fisher Scientific, Inc.). An equal
volume of 0.5 M CaCl2 was added. The
DNA/CaCl2 mixture was added to 2X HBS (500 μl; 0.28 M
NaCl, 0.05 M HEPES and 1.5 mM Na2HPO4;
optimal pH range, 7.00–7.28) and incubated at room temperature for
30 min. Subsequently, the transfection mixture was applied to
culture plates containing fresh culture media and incubated at 37°C
in a 5% CO2 humidified atmosphere. After 48 h, the
supernatants containing LvVs were collected and stored at
−80°C.
Optimal HBS pH was determined for the
production of LvVs in HEK 293T cells
To empirically determine the optimal pH of HBS for
use in the CaPO4-mediated transfections, trial
experiments (pH range, 7.0–7.28) were conducted. Following the
infection of NIH 3T3 cells (described below), the LvVs produced
from the transfected cells was quantified using the direct
titration method.
An appropriate number of NIH 3T3 cells were seeded
in a culture plate and incubated at 37°C in a 5% CO2
humidified atmosphere. After 24 h, all of the culture medium was
removed and virus medium containing polybrene was added to the
well. The culture plate was centrifuged twice at room temperature
and incubated at 37°C in a 5% CO2 humidified
atmosphere.
Titration of LvV stocks
Direct titration method
NIH 3T3 cells (2×105 cells/ml) were
trypsinized and resuspended in DMEM containing 10% FBS and
polybrene was added to a final concentration of 8 μg/ml. The NIH
3T3 cells (1 ml) and viral supernatant (5–50 μl) were transferred
into a 3 ml snap-top tube and mixed. The cell solution containing
the virus was immediately seeded in individual wells of six-well
plates and incubated at 37°C in a 5% CO2 humidified
atmosphere. After 24 h, each well was supplemented with 1 ml fresh
culture media. Three days after infection, the NIH 3T3 cells were
trypsinized and the number of fluorescence-positive cells was
determined using flow cytometry (Epics® XL™ flow
cytometer; Beckman Coulter, Inc., Mississauga, ON, Canada).
Delayed titration method
NIH 3T3 cells (2×105 cells/well) were
seeded in six-well plates and incubated at 37°C in a 5%
CO2 humidified atmosphere. After 24 h, the culture
medium was removed and 1 ml fresh DMEM containing 10% FBS, 8 μg/ml
polybrene and 5–50 μl viral supernatant were added to each well.
After 24 h, each well was supplemented with 1 ml fresh culture
medium. Three days after infection, the NIH 3T3 cells in each well
were trypsinized and the number of fluorescence-positive cells was
determined using flow cytometry (Epics XL flow cytometer; Beckman
Coulter, Inc.).
Titer formula
The following formula was used to convert the
percentage of eGFP-expressing cells for a specific dilution into
TU: TU/ml =[(% of cells expressing eGFP/100) × total number of NIH
3T3 cells at time of infection/volume of virus stock added (ml)
(23).
Flow cytometry
For immunostaining, cells were trypsinized and
washed twice with 1X PBS. After determining cell number (Vi-Cell XR
cell viability analyzer; Beckman Coulter, Inc.),
0.5–1×106 cells were resuspended in 1 ml cold PBS or
100% formalin, protected from light and measured using an Epics XL
flow cytometer (Beckman Coulter, Inc.) immediately or stored at 4°C
for future use. A total of 1×104–1×105 events
were analyzed in each sample and the resulting data were analyzed
using Summit software (version 4.3; Dako North America, Inc.,
Carpinteria, CA, USA).
Statistical analyses
Statistical analyses were performed using GraphPad
Prism statistical software (GraphPad Software, Inc., La Jolla, CA,
USA). Data was expressed as the mean ± standard error of the mean
of three independent experiments performed in triplicate. The
probability of significant differences between two groups or
multiple groups was determined using Student’s t-test or analysis
of variance, respectively. P<0.05 was considered to indicate a
statistically significant difference.
Results
Optimization of lentivirus
production
Although various commercial transfection reagents
are available, the selection of a suitable transfection system was
vital for the success of the present study. Virus production in
serum-free conditions is ideal due to the possible future uses of
virus purification and concentration. The key to successful LvV
production is the efficient transfection of HEK 293T cells. For
LvVs expressing fluorescent reporters, transfection efficiencies
can be easily determined by the direct analysis of fluorescence in
the transfected HEK 293T cells. Although more labor-intensive, the
analysis of LvV production by viral titration using flow cytometry
allows for a more precise determination of the optimal transfection
method, choice of culture medium and timing of LvV harvest that
cannot be adequately determined by looking solely at the
transfection efficiencies of the HEK 293T cells. Therefore, prior
to optimizing the transfection methods for the production of LvV,
two titration methods were investigated.
Comparison of different titration methods
of LvV stocks
Two methods were evaluated for the determination of
the LvV titer produced from the transfected HEK 293T cells. The
first method (direct titration method) was performed as follows:
Vector stock (5 μl) was immediately added to 1 ml culture medium
containing 2×105 freshly trypsinized NIH 3T3 cells
suspension in a six-well plate. After 24 h, an additional 1 ml
culture medium was added to each well. The second method (delayed
titration method) was performed as follows: NIH 3T3 cells
(2×105) were plated in each well of a six-well plate.
After 24 h, the media was removed and replaced with 1 ml culture
medium containing 5 μl vector stock. After 24 h, an additional 1 ml
culture medium was added to each well of the six-well plates. Three
days after transduction of the NIH 3T3 cells, the percentage of
eGFP-expressing NIH 3T3 cells was measured using flow cytometry and
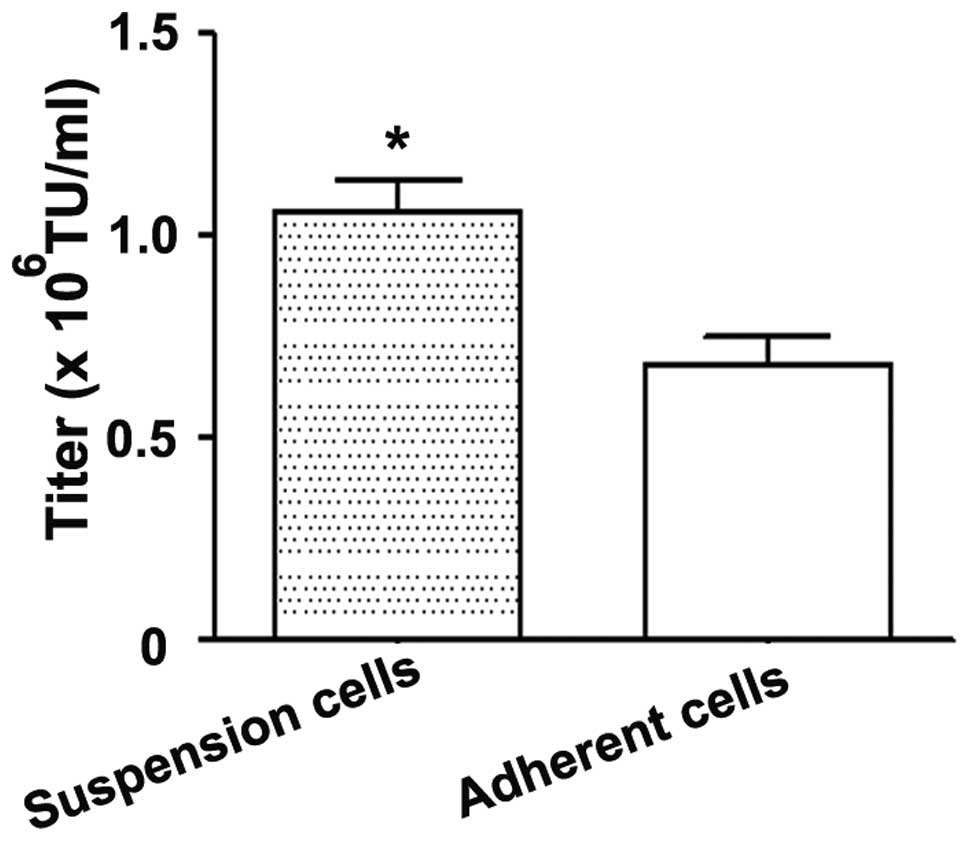
titers were calculated. As demonstrated by Fig. 1, the titer produced by direct
titration was significantly higher than that obtained by delayed
titration, which involved infected cells which had been plated 24 h
earlier. These results indicated that the direct titration method,
which is faster and simpler to perform, is a sensitive and
convenient method for viral titration.
Effect of different culture media and
culture durations on the efficiency of PEI-mediated
transfection
In the initial production trial, HEK 293T cells were
seeded at a density of 1×107 cells/10-cm plate to
produce LvVs. To quantify the amount of LvVs produced, the direct
titration method was performed by adding 50 μl viral supernatant to
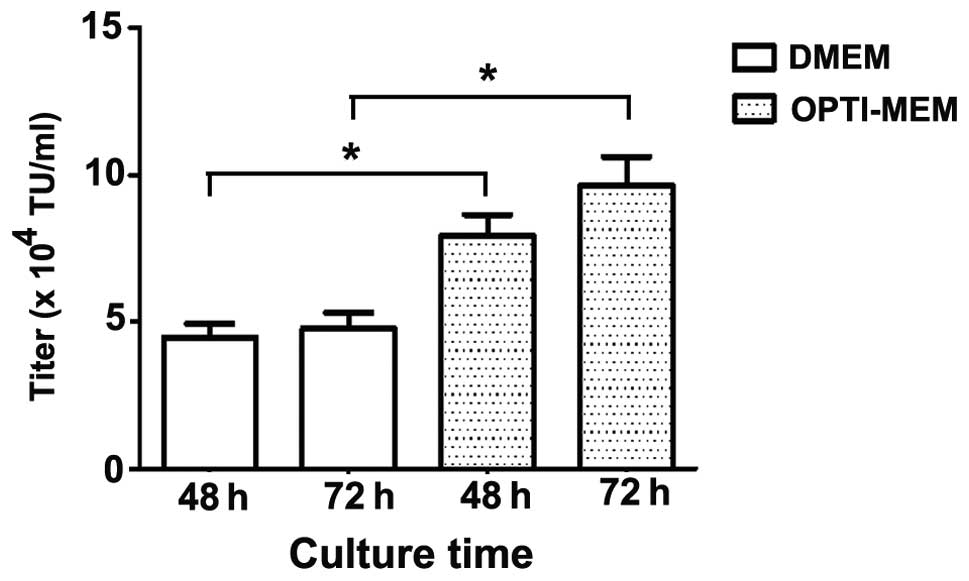
each well containing NIH 3T3 cells. As demonstrated in Fig. 2, using serum-free Opti-MEM as the
culture medium produced a higher titer compared with using DMEM,
regardless of whether LvVs were harvested 48 or 72 h after
transfection. Notably, the titers of LvV were not significantly
different at 48 and 72 h post-transfection in the Opti-MEM or DMEM
groups. These results indicated that serum-free Opti-MEM may be
used directly to produce LvVs and that LvVs in the culture medium
can be harvested 48 h after transfection, increasing the speed of
the process.
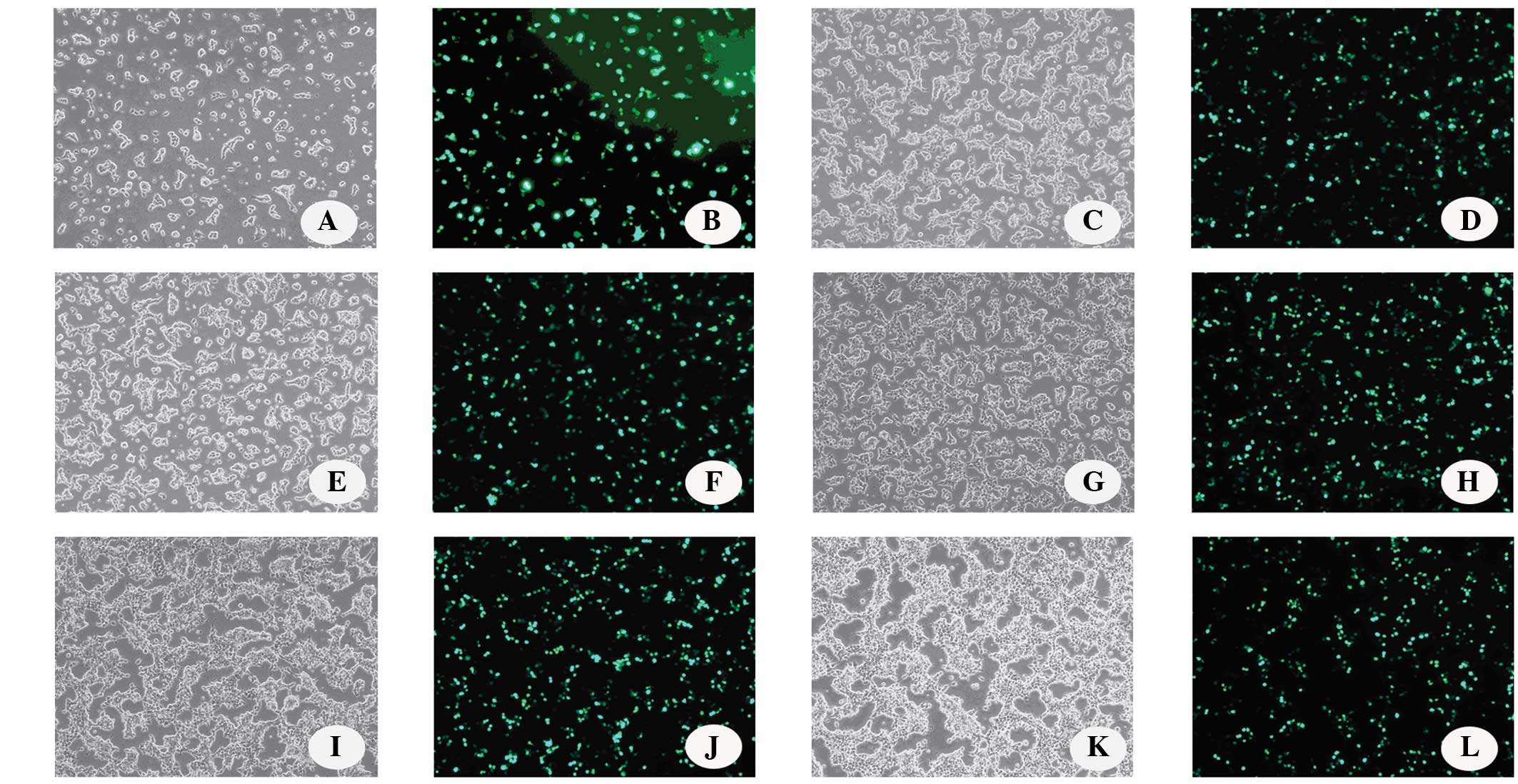
Influence of cell density on the
efficiency of PEI-mediated transfection
The optimal cell density for LvV production in 10-cm
plates was investigated. Transient transfections were performed at
low and high cell densities, from 5×106 to
40×106 cells/10-cm plate and the eGFP-expressing cells
were observed under a fluorescent microscope (Fig. 3). Although the cells could achieve a
density of >40×106 cells/10-cm plate, the percentage
of eGFP-expressing cells did not increase beyond this point when
increasing the cell density during the transient transfection.
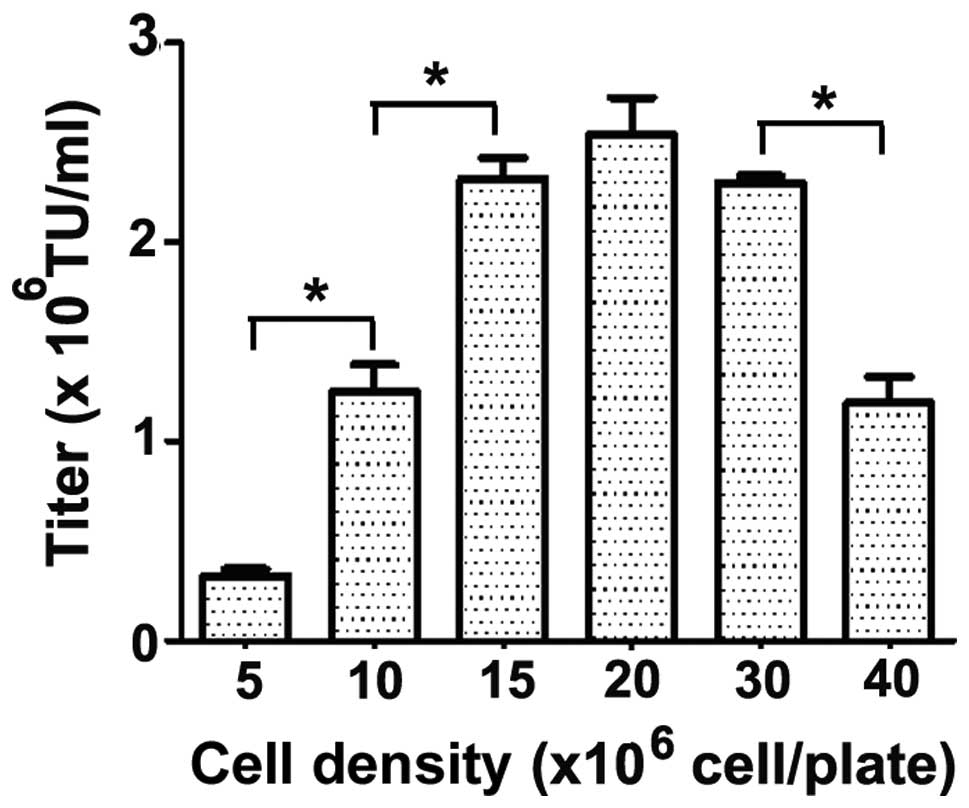
As demonstrated in Fig.
4, vector titers were dependent on cell density and a
significant increase in vector titers was achieved by increasing
the cell density from 10×106 cells/10-cm plate to
15×106 cells/10-cm plate. However, increasing the cell
density to ≥20×106 cells/10-cm plate did not
significantly increase the titer volume. Thus, 15×106
cells/10-cm plate was selected as the optimum cell density for the
production of LvVs in the subsequent experiments.
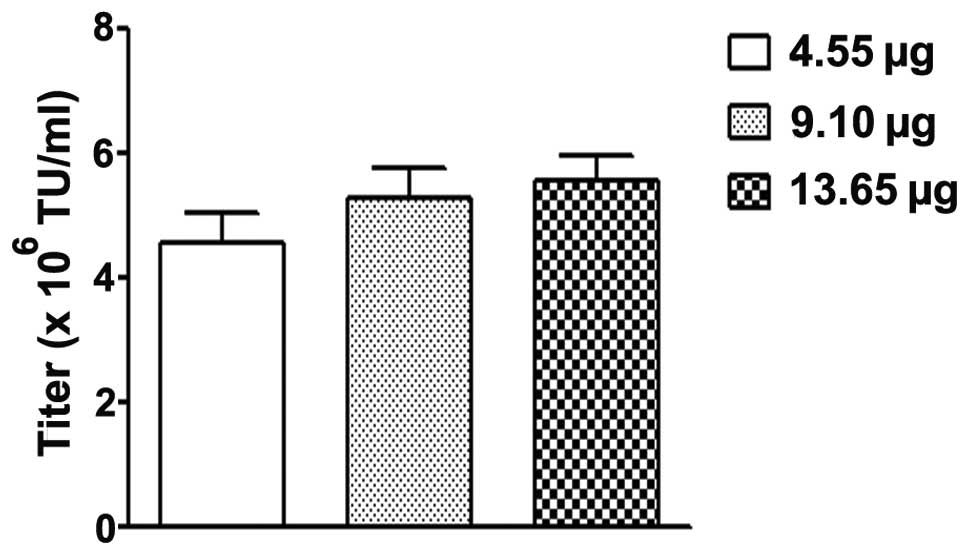
Optimization of DNA quantity for
PEI-mediated LvV production
Based on the results of the previous experiments,
Opti-MEM medium and a HEK 293T cell density of 15×106
cells/10-cm plate was selected to investigate the effects of
altered DNA concentrations on the titers of LvV produced. The
plasmid DNA solution (containing 4.55, 9.10 or 13.65 μg plasmid
DNA) was prepared as described above. As demonstrated in Fig. 5, the LvV titer was not significantly
different when different plasmid DNA concentrations were used. This
indicates that the DNA concentration is not a key factor in the
present experiment; thus, the DNA concentration could be
standardized for future experiments (4.55 μg plasmid DNA mixture
mixed with 150 mM NaCl; final volume, 380 μl).
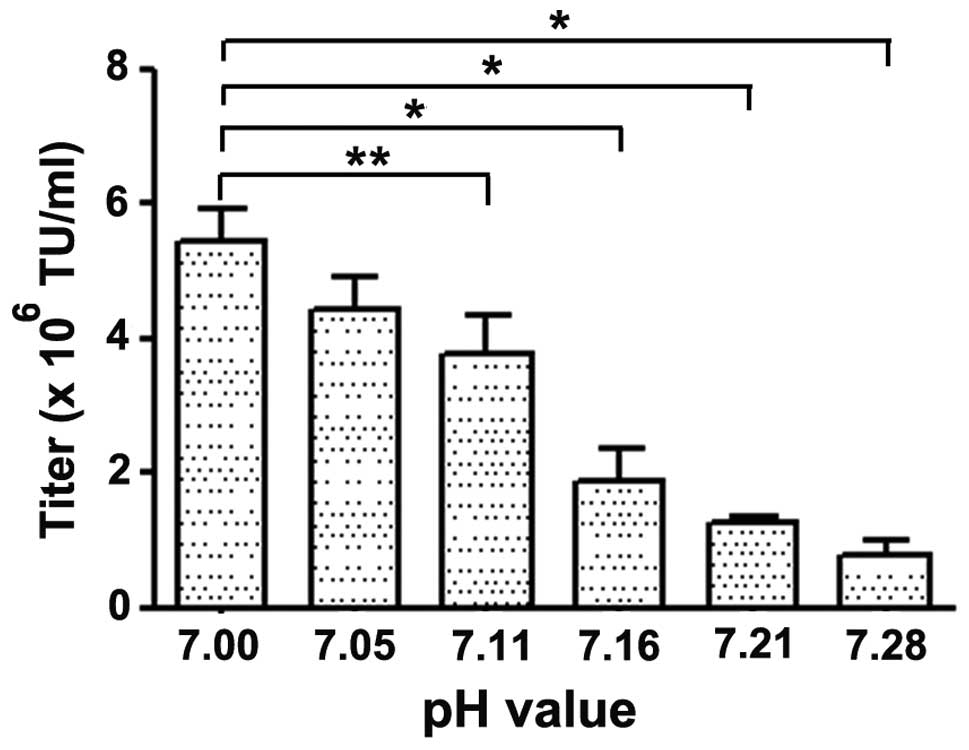
Optimization of the pH for LvV production
using CaPO4
The pH of HBS was critical during
CaPO4-mediated transient transfections. In the direct
titration method, 7.00 and 7.05 were identified to be the optimum
pH of HBS for CaPO4-mediated transient transfection
(Fig. 6). These results indicated
that the preferred HBS pH for LvV production using CaPO4
is 7.00. Both the direct observation of the percentage of
eGFP-expressing HEK 293T cells by flow cytometry and the
determination of LvV titers confirmed the optimal pH of HBS to be
~7.00. It was noted, however, that an HBS pH of 7.28 was effective
at transfecting the HEK 293T cells based on direct flow cytometry,
but produced a markedly lower LvV titer compared with at the
optimal pH of 7.00.
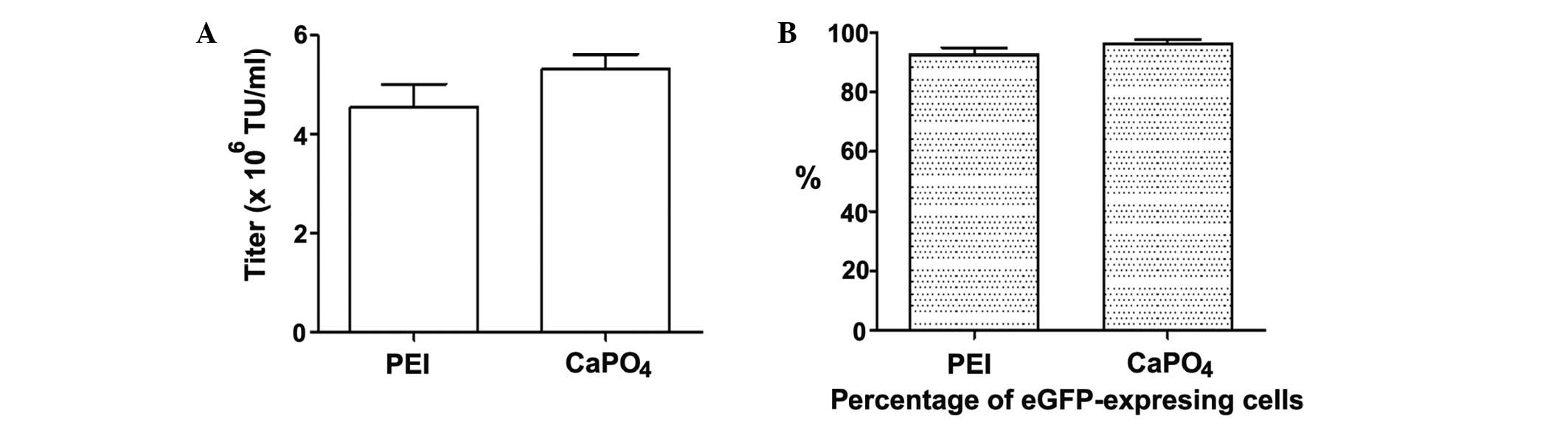
Comparison of LvV production using PEI-
and CaPO4-mediated transient transfection
The LvV titers produced by PEI- or
CaPO4-mediated transfection were compared following the
transfection of HEK 293T cells. As demonstrated in Fig. 7A, the LvV titers produced were not
significantly different between the PEI- and
CaPO4-mediated transfection methods.
The percentages of eGFP-expressing cells 48 h after
transfection were determined by fluorescent microscopy observation
of the culture plates or by analysis using a flow cytometer. No
observable difference was identified in the proportion of
eGFP-expressing cells between the PEI- and
CaPO4-mediated transient transfection culture plates.
Flow cytometry data confirmed that 92.25±2.80% of HEK 293FT cells
transfected with PEI expressed eGFP, whereas 96.15±1.44% of the
CaPO4-transfected cells were eGFP-positive. The
percentages of eGFP fluorescent cells in the PEI- and
CaPO4-transfected groups were not statistically
different (Fig. 7B).
Discussion
In the present study, the protocols for LvV
production using PEI-mediated transient transfection were
optimized. Vector titers were increased almost 100-fold after using
these protocols and were similar to those achieved using
CaPO4-mediated transient transfection. The protocol was
simple, pH-independent, economic, reproducible and consistent.
Generally, two different methods are used to produce
LvVs: The delayed titration method (which involves the development
of stable vector packaging cell lines) and the transient titration
method. Although several stable vector packaging cell lines have
been generated (24), their use has
various limitations. Firstly, the development of a stable vector
packaging cell line requires a relatively long period of selection
and characterization (25).
Secondly, transgenes or vector components may carry toxicities that
are not compatible with the development of a stable packaging cell
line (26). Additionally, for each
desired vector pseudotype, a new packaging cell line must be
developed and, finally, stable packaging cell lines often produce
relatively low vector titers and lack stability over prolonged
culture periods, limiting their long-term applications (10). As a consequence, transient
transfections of plasmids constitute a faster and simpler approach
to LvV production and avoid the time-consuming development of
stable packaging cell lines (27).
In addition, various transgenes and envelope glycoproteins can
easily be substituted in the transient transfection vector systems
(28). Thus, the production of LvVs
by transient transfection remains the most widely used technique
and has been applied in the present study.
While the transient transfection method was used in
all of the experiments in the present study, it was important to
standardize a convenient system for the routine production of LvVs.
PEI-mediated transient transfection, a simple and effective method,
was investigated for the transient transfection of HEK 293T cells
for the efficient production of LvV.
In the present study, various conditions, including
the type of medium, harvest period, cell density, amount of plasmid
DNA and titer method were evaluated and optimized to achieve the
most favorable PEI-mediated transient transfection protocol. The
resultant protocol significantly increased LvV titers by almost
100-fold compared with the non-optimized conditions. Furthermore,
vector titers similar to those achieved using the
CaPO4-mediated transient transfection method were
obtained in the serum and protein supplement-free Opti-MEM medium.
In addition to the advantages of using serum-free media, the
PEI-mediated transfection method is simpler than the
CaPO4 method in that the solutions are simple to
prepare, not dependent on pH and transfected cells do not require
their medium to be changed 24 h after transfection. Also, the
PEI-mediated method requires approximately ten-fold less DNA to
achieve similar amounts of LvV production to the
CaPO4-mediated method. In the present study,
PEI-mediated transient transfection demonstrated a high degree of
reproducibility and consistency in its LvV production compared with
CaPO4-mediated transfection. For example, the pH of the
HBS used in the CaPO4-mediated method changes during
storage, making it necessary to periodically re-prepare the buffer
using the abovementioned empirical approach. Therefore, the use of
the PEI-mediated transfection method simplifies the transfection
procedure, significantly reduces the quantity of plasmid DNA and
shortens the time required for LvV production.
Although the present study considered various
factors during the optimization of PEI-mediated transient
transfection for LvV production, there are numerous other
parameters that could also be examined. For example, the ratio of
PEI nitrogen to DNA phosphorous (N/P ratio) (29) and the quantity of DNA (22) have previously been demonstrated as
important parameters for transient transfection. The PEI volume per
plate used in the present study was calculated based on the
protocol previously described by Kuroda et al (22). However, the PEI volume per plate may
vary, perhaps in accordance with the equation described by Reed
et al (13). The present
study determined that 4.55 μg DNA mixture was adequate for the
transfection of 1.5×107 cells (0.3 μg DNA/106
cells). This quantity is similar to that described in previous
studies where 0.4–0.6 μg total DNA/106 cells was used
(22,30). Additional optimization of the amount
of DNA used for PEI-mediated transfection could possibly be
undertaken.
The use of different cell densities during
PEI-mediated transient transfection resulted in significantly
different titers (22) and the
optimal cell density varied when different types of vectors were
used. Based on the results obtained by Kuroda et al
(22), LvV titers were relatively
unaffected by 1–2×107 HEK 293T cells/15-cm plate and the
highest vector titers were obtained at a cell density of
1×107 cells/15-cm plate. In the initial experiments of
the present study, relatively lower titers were obtained when the
HEK 293T cell density was 1×107 cells/10-cm plate. This
data confirms that the cell density is one of the defining
conditions in optimizing the process of LvV production.
Furthermore, the present study did not evaluate the
effect of the addition of nutrients on the efficiency of virus
production. For example, the addition of peptones has been reported
to lead to superior transfection efficiencies using PEI-mediated
transfection (31–32).
The production of LvV in serum-free medium has
previously been described (22). In
two related studies, the PEI-mediated transfection method for LvV
production was described, using Opti-MEM reduced serum medium
(15) and serum-free, chemically
defined (FreeStyle™) medium (12).
By contrast, the present study used DMEM/high glucose and
serum-free Opti-MEM reduced serum medium to directly culture HEK
293T cells. In previous studies, the LvV in the culture medium were
typically harvested 48 h after transfection (13,22,33).
Consistent with this, the present study identified that the optimal
conditions of LvV production involved PEI-mediated transient
transfection, serum-free medium and a 48-h incubation.
Although HEK 293T cells have been widely used for
the production of LvV, including in the present study, HeLa cells
and XDC293 cells have also been used as packaging cell lines for
the production of recombinant viruses (13). In the present protocol, the titers
of infective LvP were assessed by their ability to transfer the
eGFP gene to the NIH 3T3 cell. This was similar to a study by
Salmon and Trono (7), who
recommended that the optimization of transfection protocols use a
plasmid encoding GFP when establishing vector production procedures
(7). Different LvV titration
methods, including the determination of transgenic mRNA levels,
correlate well with TU and can be used for the functional titration
of non-fluorescent transgenes (34). Similarly, the quantification of LvV
in the absence of fluorescent markers can be determined by
quantitative polymerase chain reaction. The present study used
NIH3T3 cells to titer LvV due to their restricted ability to infect
only murine cells; however, HeLa cells have also been used as
target cells for measuring titers of LvV pseudotyped with the
vesicular stomatitis virus (VSV) G glycoprotein, allowing the
infection of human cells (7).
Although not all LvV titration methods were optimized, the present
study provided a simple and efficient titration method for future
use.
An additional consideration when using PEI-mediated
transient transfection are the reported toxicities (30). The efficiency and cytotoxicity
depend on the molecular weight of PEI. Smaller PEIs are
non-cytotoxic but less efficient; for example, Toledo et al
(33) obtained a high LvV titer
(~1×107 TU/ml) using branched 25 kDa PEI. Furthermore,
cross-linked PEIs have been reported to enhance gene delivery
efficiency (29). In the present
study, which used 40 kDa PEI, no cytotoxicity was observed or
evaluated; however, the vector titers were good enough for use in
future transduction experiments.
For the optimization of CaPO4-mediated
transient transfection, only the pH of HBS was optimized, as other
parameters had previously been optimized in the laboratory.
However, the titer of CaPO4-mediated transient
transfection was affected by the structure of the envelope plasmid;
the titer obtained from the envelope protein of VSV G glycoprotein
was ~10-fold higher than from the envelope protein of rabies G
glycoprotein (35). In addition,
the titer of CaPO4-mediated transient transfection was
affected by the concentration of CaPO4, the time of
complex formation (36) and the
cell cycle (37–38).
In the present study, similar titers of LvV were
produced from PEI- and CaPO4-mediated transient
transfection. Similarly, although Toledo et al (33) obtained a high LvV titer using PEI,
the PEI-mediated transfection method did not demonstrate a higher
titer compared with the CaPO4-mediated transfection
method. By contrast, for the production of recombinant AVVs in
HEK293 and HeLa cells, linear PEI was a better transfection reagent
than CaPO4 (13). The
vector titers achieved by transient transfection in the present
study were relatively low compared with the typical titers achieved
in previous reports (106–107 TU/ml) (22,30).
This may have been due to the choice, for biosafety considerations,
to pseudotype the LvV with the ecotropic envelope protein. The use
of this envelope protein has consistently been demonstrated to
result in lowered titers when compared with the VSV G glycoprotein
(22,33).
Although numerous other factors may be optimized to
improve LvV production, it is apparent that the PEI-mediated
transfection method is the most cost-effective option (19). In addition to improved transfection
efficiencies, LvV titers may be improved by the processing of cell
culture supernatants to purify and concentrate the virus using
serum-free media (22), physical
concentration (11),
ultracentrifugation (28),
HYPERFlask® vessels (39) or filtration approaches (40), such as diafiltration (15). Although the LvV produced in the
present study was not purified, the use of the PEI-mediated
transfection method and protein-free media should allow this route
to be pursued if required.
In conclusion, the present study developed,
described and optimized a scalable process for the production of
LvV by PEI-mediated transfection. Interest in the use of LvV for
human gene therapy is high and it has even undergone phase I
clinical trials to investigate its possible use in the treatment of
human immunodeficiency virus (41).
However, large-scale LvV production remains a challenge for
clinical applications. Thus, additional developments in LvV
production and purification strategies will be essential to improve
the cost-effectiveness of LvV use in clinical studies.
Acknowledgements
The present study was supported by grants from the
Natural Science Fund Program of Guangxi Province (grant nos.
2011GXNSFC018020 and 2012GXNSFAA053163).
References
|
1
|
Quinonez R and Sutton RE: Lentiviral
vectors for gene delivery into cells. DNA Cell Biol. 21:937–951.
2002. View Article : Google Scholar
|
|
2
|
Vigna E and Naldini L: Lentiviral vectors:
excellent tools for experimental gene transfer and promising
candidates for gene therapy. J Gene Med. 2:308–316. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Segura MM, Garnier A, Durocher Y, Ansorge
S and Kamen A: New protocol for lentiviral vector mass production.
Methods Mol Biol. 614:39–52. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shin KJ, Wall EA, Zavzavadjian JR, et al:
A single lentiviral vector platform for microRNA-based conditional
RNA interference and coordinated transgene expression. Proc Natl
Acad Sci USA. 103:13759–13764. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coleman JE, Huentelman MJ, Kasparov S, et
al: Efficient large-scale production and concentration of
HIV-1-based lentiviral vectors for use in vivo. Physiol Genomics.
12:221–228. 2003.
|
|
6
|
Kosaka Y, Kobayashi N, Fukazawa T, et al:
Lentivirus-based gene delivery in mouse embryonic stem cells. Artif
Organs. 28:271–277. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salmon P and Trono D: Production and
titration of lentiviral vectors. Curr Protoc Neurosci.
37:4.21.1–4.21.24. 2006.
|
|
8
|
Lee JH and Welsh MJ: Enhancement of
calcium phosphate-mediated transfection by inclusion of adenovirus
in coprecipitates. Gene Ther. 6:676–682. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mochizuki H, Schwartz JP, Tanaka K, Brady
RO and Reiser J: High-titer human immunodeficiency virus type
1-based vector systems for gene delivery into nondividing cells. J
Virol. 72:8873–8883. 1998.PubMed/NCBI
|
|
10
|
Cronin J, Zhang XY and Reiser J: Altering
the tropism of lentiviral vectors through pseudotyping. Curr Gene
Ther. 5:387–398. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Segura MM, Kamen A and Garnier A:
Downstream processing of oncoretroviral and lentiviral gene therapy
vectors. Biotechnol Adv. 24:321–337. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Segura MM, Garnier A, Durocher Y, Coelho H
and Kamen A: Production of lentiviral vectors by large-scale
transient transfection of suspension cultures and affinity
chromatography purification. Biotechnol Bioeng. 98:789–799. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reed SE, Staley EM, Mayginnes JP, Pintel
DJ and Tullis GE: Transfection of mammalian cells using linear
polyethylenimine is a simple and effective means of producing
recombinant adeno-associated virus vectors. J Virol Methods.
138:85–98. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Durocher Y, Pham PL, St-Laurent G, et al:
Scalable serum-free production of recombinant adeno-associated
virus type 2 by transfection of 293 suspension cells. J Virol
Methods. 144:32–40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Geraerts M, Michiels M, Baekelandt V,
Debyser Z and Gijsbers R: Upscaling of lentiviral vector production
by tangential flow filtration. J Gene Med. 7:1299–1310. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boussif O, Lezoualc’h F, Zanta MA, et al:
A versatile vector for gene and oligonucleotide transfer into cells
in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA.
92:7297–7301. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pham PL, Kamen A and Durocher Y:
Large-scale transfection of mammalian cells for the fast production
of recombinant protein. Mol Biotechnol. 34:225–237. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Derouazi M, Girard P, Van Tilborgh F, et
al: Serum-free large-scale transient transfection of CHO cells.
Biotechnol Bioeng. 87:537–545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Durocher Y, Perret S and Kamen A:
High-level and high-throughput recombinant protein production by
transient transfection of suspension-growing human 293-EBNA1 cells.
Nucleic Acids Res. 30:E92002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao DS, Li L, Garson K and Vanderhyden BC:
OPCML gene transferred by recombinant lentiviruses in vitro and its
inhibition to ovarian cancer cells. Zhonghua Fu Chan Ke Za Zhi.
41:333–338. 2006.(In Chinese). PubMed/NCBI
|
|
21
|
Jounai N, Okuda K, Kojima Y, et al:
Contribution of the rev gene to the immunogenicity of DNA vaccines
targeting the envelope glycoprotein of HIV. J Gene Med. 5:609–617.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuroda H, Kutner RH, Bazan NG and Reiser
J: Simplified lentivirus vector production in protein-free media
using polyethylenimine-mediated transfection. J Virol Methods.
157:113–121. 2009. View Article : Google Scholar
|
|
23
|
Kahl CA, Marsh J, Fyffe J, Sanders DA and
Cornetta K: Human immunodeficiency virus type 1-derived lentivirus
vectors pseudotyped with envelope glycoproteins derived from Ross
River virus and Semliki Forest virus. J Virol. 78:1421–1430. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kafri T, van Praag H, Ouyang L, Gage FH
and Verma IM: A packaging cell line for lentivirus vectors. J
Virol. 73:576–584. 1999.
|
|
25
|
Ni Y, Sun S, Oparaocha I, et al:
Generation of a packaging cell line for prolonged large-scale
production of high-titer HIV-1-based lentiviral vector. J Gene Med.
7:818–834. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sinn PL, Sauter SL and McCray PB Jr: Gene
therapy progress and prospects: development of improved lentiviral
and retroviral vectors - design, biosafety, and production. Gene
Ther. 12:1089–1098. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mitta B, Rimann M and Fussenegger M:
Detailed design and comparative analysis of protocols for optimized
production of high-performance HIV-1-derived lentiviral particles.
Metab Eng. 7:426–436. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sena-Esteves M, Tebbets JC, Steffens S,
Crombleholme T and Flake AW: Optimized large-scale production of
high titer lentivirus vector pseudotypes. J Virol Methods.
122:131–139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thomas M, Ge Q, Lu JJ, Chen J and Klibanov
AM: Cross-linked small polyethylenimines: while still nontoxic,
deliver DNA efficiently to mammalian cells in vitro and in vivo.
Pharm Res. 22:373–380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun X, Hia HC, Goh PE and Yap MG:
High-density transient gene expression in suspension-adapted 293
EBNA1 cells. Biotechnol Bioeng. 99:108–116. 2008. View Article : Google Scholar
|
|
31
|
Pham PL, Perret S, Doan HC, et al:
Large-scale transient transfection of serum-free suspension-growing
HEK293 EBNA1 cells: peptone additives improve cell growth and
transfection efficiency. Biotechnol Bioeng. 84:332–342. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pham PL, Perret S, Cass B, et al:
Transient gene expression in HEK293 cells: peptone addition
posttransfection improves recombinant protein synthesis. Biotechnol
Bioeng. 90:332–344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Toledo JR, Prieto Y, Oramas N and Sánchez
O: Polyethylenimine-based transfection method as a simple and
effective way to produce recombinant lentiviral vectors. Appl
Biochem Biotechnol. 157:538–544. 2009. View Article : Google Scholar
|
|
34
|
Geraerts M, Willems S, Baekelandt V,
Debyser Z and Gijsbers R: Comparison of lentiviral vector titration
methods. BMC Biotechnol. 6:342006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reiser J: Production and concentration of
pseudotyped HIV-1-based gene transfer vectors. Gene Ther.
7:910–913. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sakoda T, Kasahara N, Kedes L and Ohyanagi
M: Calcium phosphate coprecipitation greatly enhances transduction
of cardiac myocytes and vascular smooth muscle cells by lentivirus
vectors. Exp Clin Cardiol. 12:133–138. 2007.
|
|
37
|
Trobridge G and Russell DW: Cell cycle
requirements for transduction by foamy virus vectors compared to
those of oncovirus and lentivirus vectors. J Virol. 78:2327–2335.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
O’Rourke JP, Newbound GC, Kohn DB, Olsen
JC and Bunnell BA: Comparison of gene transfer efficiencies and
gene expression levels achieved with equine infectious anemia
virus- and human immunodeficiency virus type 1-derived lentivirus
vectors. J Virol. 76:1510–1515. 2002. View Article : Google Scholar
|
|
39
|
Kutner RH, Puthli S, Marino MP and Reiser
J: Simplified production and concentration of HIV-1-based
lentiviral vectors using HYPERFlask vessels and anion exchange
membrane chromatography. BMC Biotechnol. 9:102009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koldej R, Cmielewski P, Stocker A, Parsons
DW and Anson DS: Optimisation of a multipartite human
immunodeficiency virus based vector system; control of virus
infectivity and large-scale production. J Gene Med. 7:1390–1399.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McGarrity GJ, Hoyah G, Winemiller A, et
al: Patient monitoring and follow-up in lentiviral clinical trials.
J Gene Med. 15:78–82. 2013. View Article : Google Scholar : PubMed/NCBI
|