Introduction
Pituitary adenomas are a relatively common type of
intracranial neoplasm, however, the mechanisms underlying its
pathogenesis have yet to be fully elucidated. Genetic alterations,
such as missense mutations in the gene-encoding α-subunit of G
protein (gsα) or the MEN1 gene in multiple endocrinologic
neoplasia syndrome 1 (MEN1) have been identified in pituitary
adenomas (1–5).
Gsα mutations have been identified in growth hormone
(GH)-secreting pituitary adenomas and non-functioning pituitary
adenomas. These mutations include the replacement of arginine by
cysteine, serine or histidine in codon 201 of exon 8, or the
replacement of glutamine by arginine or leucine in codon 227 of
exon 9 (1,6,7).
Furthermore, a previous study reported that the frequency of gsα
mutations in patients with GH-secreting pituitary adenomas ranged
between 4.4 and 43% (2,3).
Patients with MEN1 are predisposed to developing
tumors of the parathyroid, pancreas and pituitary gland. The
disease is caused by inactivating mutations in MEN1, a
putative tumor suppressor gene, which has been localized to
chromosome 11q13 by genetic mapping studies (8). Loss of heterozygosity has been
identified in MEN1-associated pituitary adenomas and in
10–18% of sporadic pituitary adenoma patients, indicating the
involvement of MEN1 (9).
Furthermore, a significant proportion of sporadic pituitary tumors
harboring deletions map to the critically deleted region of the
MEN1 gene (4). The present
study describes the case of a female patient with a coexisting
GH-producing pituitary tumor, papillary thyroid carcinoma and
subcutaneous fibroma. Despite surgery, Gamma-Knife®
radiosurgery and octreotide acetate treatment of the GH-producing
pituitary tumor, the patient’s GH levels remained elevated, with no
evidence of residual pituitary tumor on a computed tomography scan.
Thus, the germinal mutations in the MEN1 and gsα genes were
analyzed to investigate the molecular pathology of the tumors.
Written informed consent was obtained from the patient.
Patient and methods
Case report
In September 2012, a 39-year-old female presented to
the Department of Endocrinology, The First Hospital of Lanzhou
University (Lanzhou, China) with progressive enlargement of the
hands, feet and lips for 12 years. In June 2000, the patient
presented with typical manifestations of acromegaly, including
progressive enlargement of the hands, feet, lip and tongue,
pachylosis and sleep apnea. In April 2007, the patient underwent
surgery to remove a nodule in the left shoulder, which was
histopathologically diagnosed as a subcutaneous fibroma. In March
2009, magnetic resonance imaging revealed a pituitary macroadenoma
with a GH level of >40 ng/ml. The patient underwent a pituitary
adenoma resection via the single nostril transsphenoidal approach,
and subsequent pathological analysis of the tumor was consistent
with a pure, densely granulated, GH-producing pituitary adenoma
(somatotropinoma). In addition, in August 2009 and March 2012, the
patient underwent Gamma-Knife radiosurgery for the treatment of
residual tumor detected by a computed tomography scan and for
persistent acromegaly with GH levels of >20 ng/ml, respectively.
However, three months after Gamma-Knife treatment, the patient’s GH
levels increased to 21.2 ng/ml.
Finally, in November 2011, cervical ultrasonography
revealed a 1.3×1.8-cm, irregular-shaped nodule on the left
posterior lobe of the thyroid and a 1.1×0.8-cm, irregular-shaped
nodule on the right lobe of the thyroid. The patient therefore
underwent a total thyroidectomy and ipsilateral level VI lymph node
dissection. Subsequent histopathological analysis revealed a left
thyroid papillary carcinoma, which was negative for RET expression,
therefore, levothyroxine was administered orally at a dose of 150
μg/day following thyroid surgery.
In September 2012, a physical examination indicated
a blood pressure of 110/68 mmHg, and acromegaly manifesting as
prognathism, a prominent forehead, flat nose and macroglossia.
Furthermore, mild anemia without jaundice and no lymphadenopathy
were observed.
The results of the laboratory analyses that were
conducted are summarized in Table
I. Thyroid function tests indicated the following levels:
Thyroid-stimulating hormone, 0.229 μIU/ml (normal range, 0.55–4.78
μIU/ml); triiodothyronine (T3), 1.02 ng/ml (normal
range, 0.60–1.81 ng/ml); thyroxine (T4), 10.50 μg/dl
(normal range, 4.50–10.90 μg/dl); free(F)T3, 3.33 pg/ml
(normal range, 2.3–4.2 pg/ml); FT4, 1.73 ng/dl (normal
range, 0.89–1.76 ng/dl); and thyroglobulin, <0.1mg/ml.
Additionally, the GH level was 23.94 ng/ml (nadir GH level
following glucose load of 19.57 ng/ml) and the insulin-like growth
factor level was 1103 ng/ml (normal range, 570±25 ng/ml). The sex
hormone, adrenocorticotropic hormone, cortisol and prolactin levels
were within the normal ranges, and the bone mineral density at the
calcaneus was 0.154 g/cm2 (normal range, >0.407
g/cm2) with a T-score of -4.3 (normal range,
>-1.0).
 | Table ILaboratory analysis results. |
Table I
Laboratory analysis results.
| Parameter | Value | Normal value
range |
|---|
| Hemoglobin, g/l | 87.00 | 110–150 |
| Erythrocyte,
1012/l | 2.79 | 3.5–5.0 |
| Leukocyte,
109/l | 4.44 | 4.0–10.0 |
| Folic acid,
ng/ml | 14.80 | 3.1–17.5 |
| Vitamin B12,
pg/ml | 403.90 | 211–946 |
| Serum iron,
μmol/l | 6.60 | 9.0–27.0 |
| Unsaturated iron
binding capacity, μmol/l | 72.40 | 31–51 |
| Total iron-binding
capacity, μmol/l | 79.00 | 54–77 |
| Iron saturation | 0.08 | 0.15–0.55 |
| Ferritin, ng/ml | 7.00 | 13.0–150.0 |
| Serum calcium,
mmol/l | 2.25 | 2.10–2.80 |
| Serum phosphorus,
mmol/l | 1.67 | 0.97–1.60 |
| Intact parathyroid
hormone, pg/ml | 24.70 | 14–72 |
| 2,5-hydroxy vitamin
D, nmol/l | 65.43 | 47.7–144.0 |
| Osteocalcin,
ng/ml | 41.60 | 12.8–55.0 |
| Bone-specific
alkaline phosphatase, μg/l | 17.50 | 7.3–22.4 |
Treatment and follow-up
The patient continued with life-long levothyroxine
treatment, at a dose of 175 μg/day, in addition to medication for
hypoferric anemia (50 mg iron-dextrin, three times daily) and
osteoporosis (0.25 μg calcitriol, once daily) for six months. A
computed tomography scan did not identify any evidence of residual
tumor. Monthly treatment with 20 mg octreotide acetate was
initiated. Following one month of octreotide acetate treatment, the
patient’s GH levels decreased to 4.40 ng/ml and at three months,
the GH levels were 5.47 ng/ml. Therefore, octreotide acetate
treatment was terminated, however, two months later the GH levels
increased again to 16.44 ng/ml.
Genomic DNA extraction
Peripheral venous blood (2 ml) was obtained from the
patient and 10 healthy controls. DNA was isolated from the blood
using a blood Genome DNA Extraction kit (Takara Biotechnology Co.,
Ltd., Dalian, China), dissolved in TE buffer and stored at −20°C.
The present study was approved by the Human Ethics Review Committee
of The First Hospital of Lanzhou University (Lanzhou, China) and
the investigations involving human subjects complied with the
principles outlined in the 1983 Declaration of Helsinki.
DNA amplification and mutation
detection
Exons 1–10 of the MEN1 gene were amplified by
polymerase chain reaction (PCR). All the amplified genes and the
primers used are indicated in Table
II. In addition, a 539-bp fragment, including exons 8 and 9 of
the gsα gene, was amplified by PCR using the primer sequences as
follows: Forward, 5′-GTGATCAAGCAGGCTGACTATGTG-3′ and reverse,
5′-GCTGCTGGCCACCACGAAGATGAT-3′.
| Table IIPrimers and length of the MEN1
gene amplification. |
Table II
Primers and length of the MEN1
gene amplification.
| Exon | Forward primer
(5′→3′) | Reverse primer
(5′→3′) | Length, bp | Annealing
temperature, °C |
|---|
| 2A |
TCCCTCCCCCGGCTTGCCTT |
ACGTTGGTAGGGATGACGCG | 220 | 60 |
| 2B |
TGCTGGGCTTCGTGGAGCAT |
GAGACACCCCCTTCTCGAGG | 220 | 57 |
| 2C |
GCCCGCTTCACCGCCCAGAT |
GGAGGGTTTTGAAGAAGTGG | 230 | 58 |
| 3 |
TCATTACCTCCCCCTTCCAC |
AGGCTGGGGGGAGGGAACAA | 254 | 60 |
| 4 |
AGGGTGGGCCATCATGAGAC |
TAGCCCAGTCCTGCCCCATT | 207 | 60 |
| 5,6 |
CATAACTCTCTCCTTCGGCT |
TCTGCACCCTCCTTAGATGC | 260 | 60 |
| 7 |
GGATCCTCTGCCTCACCTCC |
GCAGGCCCTAGTAGGGGGAT | 189 | 63 |
| 8 |
AGAGACCCCACTGCTCTCACA |
GGACACAGGCTGGAGCTCC | 187 | 64 |
| 9 |
AGAGACTGATCTGTGCCCTC |
AGACCTCTGTGCAGCTGTCC | 227 | 62 |
| 10A |
ACGGGCTTGTCAGACTTTTC |
ATGCCCTTCATCTTCTCACTC | 498 | 64 |
| 10B |
GCCAGCACTGGACAAGGGCC |
CAGCAGCTCCTTCATGCCCT | 205 | 65 |
| 10C |
GGGTCCAGTGCTCACTTTCC |
CAAGCGGTCCGAAGTCCCCA | 218 | 63 |
The PCR amplification was performed in a total
volume of 50 μl containing 2 μl extracted DNA, 20 pmol of each of
the forward and reverse primers, 19 μl 2× Taq PCR Master Mix
(Takara Biotechnology Co., Ltd.) and 25 μl deionized water. The PCR
cycling conditions were as follows: Initial activation of the DNA
polymerase at 95°C for 5 min, followed by 30 cycles at 95°C for 30
min, 52–65°C for 30 min and 72°C for 45 min, and a final extension
of 72°C for 5 min. The PCR products were purified and directly
sequenced to detect gene mutations, and the sequence results were
compared with the normal sequences of the MEN1 or gsα genes
obtained from GenBank.
Results
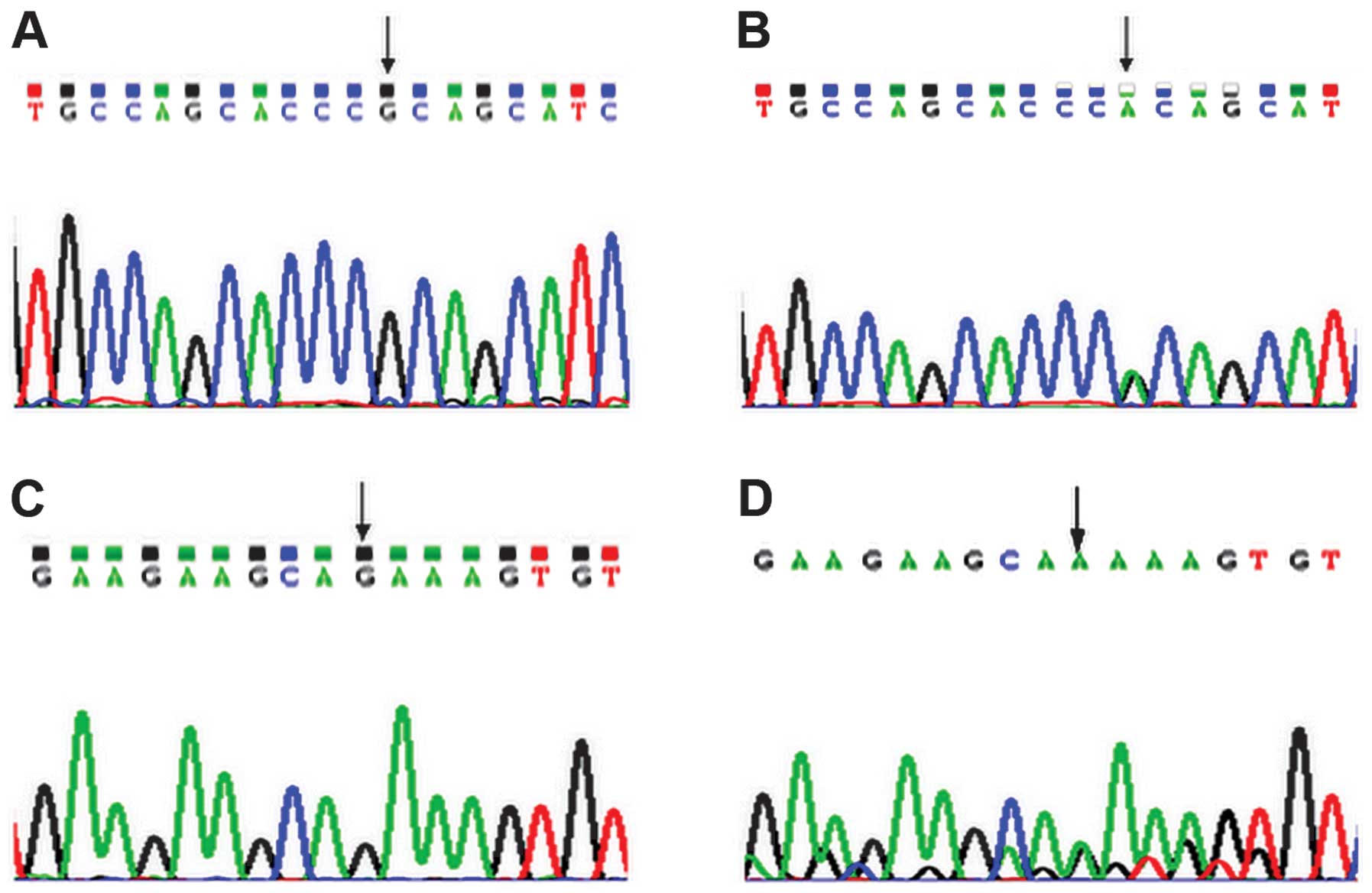
A base G was shown in healthy controls at nucleotide
7848 within exon 10 of the MEN1 gene (Fig. 1A), however, a G→A mutation was
identified in the present patient at nucleotide 7848 within exon 10
of the MEN1 gene (Fig. 1B).
The heterozygous missense mutation caused the substitution of
alanine (GCA) with threonine (ACA) at amino acid position 541
(A541T) of the MEN1 protein. The forward and reverse sequencing
results were consistent. In addition, a base G was shown in healthy
controls at nucleotide 7997 within exon 10 of the MEN1 gene
(Fig. 1C), however, the patient
demonstrated a G→A mutation at nucleotide 7997 within exon 10 of
the MEN1 gene. The mutation was synonymous, therefore, the
proline located at amino acid position 590 of the MEN1 protein was
unchanged (P590P; Fig. 1D). No
other mutation was observed in exons 8 and 9 of the gsα gene of the
patient.
Discussion
The MEN1 gene consists of 10 exons, from
which exons 2–10 encode a 610-amino acid nuclear protein known as
menin. Menin is important in the regulation of DNA transcription
and replication, cellular apoptosis, the cell cycle and the
maintenance of genome integrity (10). A MEN1 gene mutation has been
observed in patients exhibiting MEN1, an autosomal dominant genetic
disease that is characterized by the development of primary tumors
that involve two or more endocrine tissues within a single patient,
including tumors of the parathyroid gland (95% of cases),
pancreatic islets (30–80% of cases) and anterior pituitary gland
(15–90% of cases). In addition, >25% of MEN1 patients have been
reported to develop thyroid tumors, including adenomas, carcinomas
and colloid goiters (11).
Furthermore, endocrine-inactive tumors, such as lipomas,
angiofibromas and collagenomas are frequently identified in MEN1
patients (12).
In total, >400 varying germline mutations have
been identified throughout the MEN1 gene, which indicates
that such mutations are not restricted to a specific functional
domain (5). The germline mutations
identified include ~50% frameshift insertions and deletions, ~20%
nonsense mutations, ~20% missense mutations and ~7% splice-site
defects, of which the nonsense mutations, as well as many of the
frameshift insertions and deletions and the donor-splice site
mutations are loss-of-function truncating mutations involving the
menin protein. However, a definitive genotype-phenotype correlation
has yet to be established. The proposed mechanism linking
MEN1 mutations with tumor formation involves disruption of
the interactions between menin and other proteins, such as
activating protein-1 (AP-1), transcription factor Jun D or nuclear
factor-κB (NF-κB) to suppress Jun-mediated or NF-κB-mediated
transcriptional activation, and between members of the Smad family,
Smad3 and the Smad 1/5 complex, ultimately resulting in modified
cell cycle regulation and proliferation (13,14).
GH-producing tumors causing acromegaly occur in 3–6%
of MEN1 patients (15), and an
increased female-to-male ratio in MEN1 patients with pituitary
adenoma and acromegaly is observed in familiar and sporadic cases
(16). The patient in the present
study developed pituitary somatotroph adenomas and papillary
thyroid carcinoma without the other tumors commonly associated with
MEN1, such as parathyroid, intestinal pancreatic endocrine or
adrenal cortical tumors. However, genetic analysis revealed a G→A
missense mutation at nucleotide 7848 within exon 10 of the
MEN1 gene, which resulted in the substitution of alanine
with threonine at amino acid 541 (A541T) of the menin protein. The
present study proposes that the patient’s clinical manifestations
may be typical of MEN1 as opposed to pure endocrine tumors; with
the presence of subcutaneous fibroma being a specific and unusual
clinical manifestation of MEN1. Therefore, a long-term follow-up is
required to identify additional endocrine tumors associated with
MEN1.
Various studies have demonstrated the prevalence of
gsα mutations in GH-secreting pituitary adenomas varies according
to geographical location and genetic background of the population
(1–3,7),
however, the patient described in the present study demonstrated no
mutations in exons 8 and 9 of the gsα gene. Previous studies
indicated that the prevalence of gsα mutations in GH-secreting
pituitary adenomas varies by the geographical location and the
genetic background of the population (7,17).
In conclusion, the present study reported the rare
case of a patient with coexisting GH-producing pituitary tumors
causing acromegaly, papillary thyroid carcinoma and subcutaneous
fibroma. Despite pituitary surgery, Gamma-Knife radiosurgery and
octreotide acetate treatment, the patient’s GH levels were elevated
with no evidence of a residual pituitary tumor upon computed
tomography scans. Genetic analysis identified a G→A missense
mutation at nucleotide 7848 within exon 10 of the MEN1 gene,
which caused the substitution of alanine by threonine at amino acid
541 (A541T) of menin. Therefore, the pituitary adenomas and thyroid
carcinoma exhibited by the present patient may be considered as
early manifestations of MEN1 as opposed to pure endocrine tumors.
Although there is currently no evidence of other types of tumors
associated with MEN1, including parathyroid, intestinal pancreatic
endocrine or adrenal cortical tumors, long-term follow-up is
necessary.
References
|
1
|
Bezerra MG, Latronico AC and Fragoso MC:
Endocrine tumors associated to protein Gsalpha/Gi2alpha mutations.
Arq Bras Endocrinol Metabol. 49:784–790. 2005.(In Portuguese).
View Article : Google Scholar
|
|
2
|
Hosoi E, Yokogoshi Y, Hosoi E, et al:
Analysis of the Gs alpha gene in growth hormone-secreting pituitary
adenomas by the polymerase chain reaction-direct sequencing method
using paraffin-embedded tissues. Acta Endocrinol (Copenh).
129:301–306. 1993.
|
|
3
|
Kim HJ, Kim MS, Park YJ, et al: Prevalence
of Gs alpha mutations in Korean patients with pituitary adenomas. J
Endocrinol. 168:21–226. 2001. View Article : Google Scholar
|
|
4
|
Wenbin C, Asai A, Teramoto A, Sanno N and
Kirino T: Mutations of the MEN1 tumor suppressor gene in sporadic
pituitary tumors. Cancer Lett. 142:43–47. 1997. View Article : Google Scholar
|
|
5
|
Agarwal SK, Kester MB, Debelenko LV, et
al: Germline mutations of the MEN1 gene in familial multiple
endocrine neoplasia type 1 and related states. Hum Mol Genet.
6:1169–1175. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pertuit M, Barlier A, Enjalbert A and
Gérard C: Signalling pathway alterations in pituitary adenomas:
involvement of Gsalpha, cAMP and mitogen-activated protein kinases.
J Neuroendocrinol. 21:869–877. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mantovani G, Lania AG and Spada A: GNAS
imprinting and pituitary tumors. Mol Cell Endocrinol. 326:15–18.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chandrasekharappa SC, Guru SC, Manickam P,
et al: Positional cloning of the gene for multiple endocrine
neoplasia-type 1. Science. 276:404–407. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boggild MD, Jenkinson S, Pistorello M, et
al: Molecular genetic studies of sporadic pituitary tumors. J Clin
Endocrinol Metab. 78:387–392. 1994.PubMed/NCBI
|
|
10
|
Guru SC, Goldsmith PK, Burns AL, Marx SJ,
Spiegel AM, Collins FS and Chandrasekharappa SC: Menin, the product
of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA.
95:1630–1634. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pannett AA and Thakker RV: Multiple
endocrine neoplasia type 1. Endocr Relat Cancer. 6:449–473. 1999.
View Article : Google Scholar
|
|
12
|
Kawa S, Karasawa Y, Oguchi H and Furuta S:
Multiple endocrine neoplasia type I. Nihon Rinsho. 53:2702–2707.
1995.(In Japanese). PubMed/NCBI
|
|
13
|
Agarwal SK, Kennedy PA, Scacheri PC, et
al: Menin molecular interactions: insights into normal functions
and tumorigenesis. Horm Metab Res. 37:369–374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaji H, Canaff L, Lebrun JJ, Goltzman D
and Hendy GN: Inactivation of menin, a Smad3-interacting protein,
blocks transforming growth factor type β signaling. Proc Natl Acad
Sci USA. 98:3837–3842. 2001. View Article : Google Scholar
|
|
15
|
Marx S, Spiegel AM, Skarulis MC, Doppman
JL, Collins FS and Liotta LA: Multiple endocrine neoplasia type 1:
clinical and genetic topics. Ann Intern Med. 129:484–494. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vergès B, Boureille F, Goudet P, et al:
Pituitary disease in MEN type 1 (MEN1): data from the
France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab.
87:457–465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yasufuku-Takano J, Takano K, Morita K,
Takakura K, Teramoto A and Fujita T: Does the prevalence of gsp
mutations in GH-secreting pituitary adenomas differ geographically
or racially? Prevalence of gsp mutations in Japanese patients
revisited. Clin Endocrinol (Oxf). 64:91–96. 2006. View Article : Google Scholar
|