Introduction
Angiosarcoma is a rare malignant neoplasm that
affects the endothelial-type cells lining the vessel walls
(1). Overall, it accounts for
<2% of all soft-tissue sarcomas. Since the first report of the
disease in 1942 (2), extremely few
cases have been described in PubMed. Visceral sarcomas are less
common than soft-tissue and skin sarcomas, and renal involvement is
generally associated with metastasis (3). Primary renal angiosarcoma usually
affects Caucasians in the sixth and seventh decades of life. In
general, it presents with macroscopic hematuria (81%), pain in the
flank (38%) or a palpable renal mass (31%) (4). Here, we present the case of a patient
with primary renal angiosarcoma with pleuropulmonary metastasis.
The aim of this report is to provide additional information
regarding imaging techniques and the clinical behaviour of
angiosarcoma to aid with its differential diagnosis. Written
informed consent was obtained from the patient’s family.
Case report
A 57-year-old male was referred to the Gayrettepe
Florence Nightingale Hospital (Istanbul, Turkey) with right-sided
flank pain and hematuria. A physical examination revealed
tenderness in the right hypochondrium, with otherwise normal
clinical findings. The routine blood tests revealed anemia with a
hemoglobin level of 10.5 g/dl (normal range, 13–17 g/dl). The
patient underwent abdominal sonography, which identified a large
right-sided renal mass and a simple cortical cyst in the left
kidney. The remaining blood tests and tumor marker levels
[carcinoembryonic antigen, cancer antigen (CA)19-9 and CA-125] were
normal. There was no history of exposure to radiation, vinyl
chloride or thorium dioxide (also known as thorotrast).
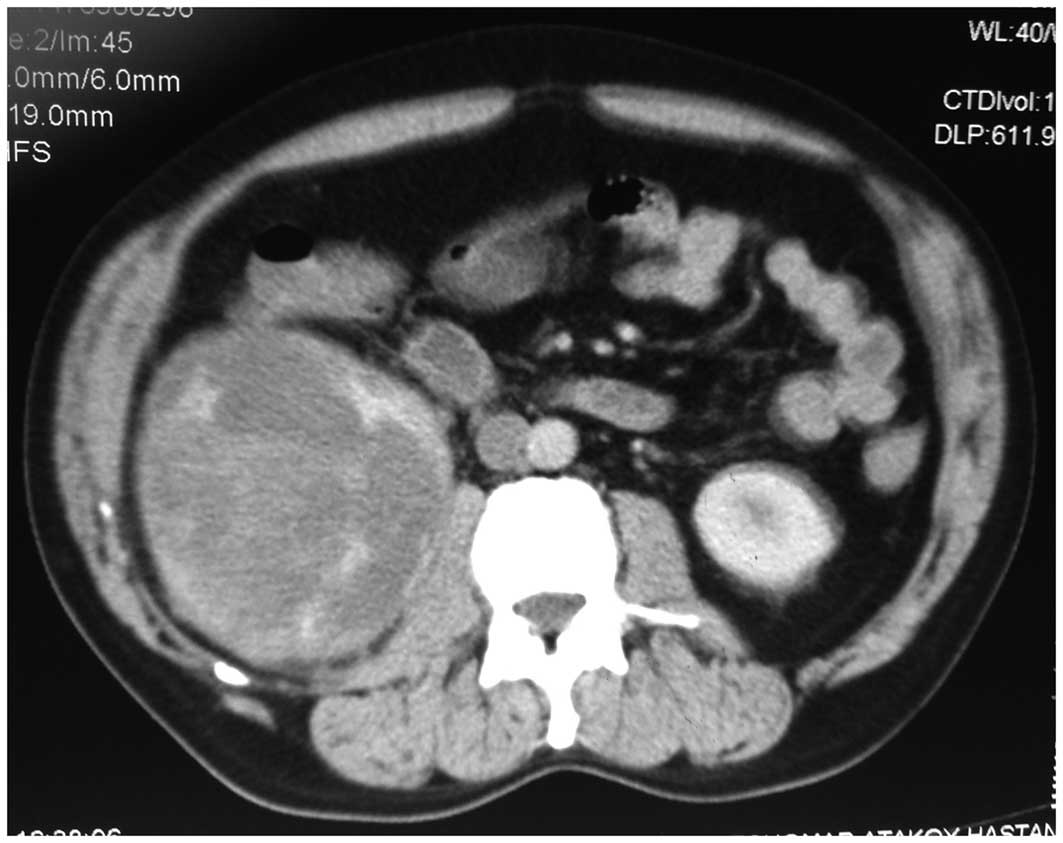
Computed tomography (CT) identified a renal mass
measuring 14×12 cm. The lesion demonstrated heterogenous peripheral
enhancement with central areas of necrosis and hemorrhage (Fig. 1). Radiological differential
diagnosis concluded that the lesion was most likely renal cell
carcinoma or a metastasis from another primary tumor. The mass was
surgically removed by radical nephrectomy. The surgical specimen
consisted of the entire right kidney, which weighed 1,080 g and
measured 18×13×10 cm, with a 14×12×9 cm mass that occupied the
majority of the tissue. The tumor was composed of spindle and
epithelioid cells with high-grade morphologies, and large, necrotic
hemorrhagic areas. Within the tumor, five mitoses per 10 high-power
fields were also observed. Normal renal tissue, measuring 7×5×4 cm,
was evident on one side of the resected specimen. There was no
evidence of thrombosis in the renal vein. Immunohistochemical
findings revealed that the neoplastic cells were highly positive
for cluster of differentiation (CD)31 and CD34, which supported the
diagnosis of primary renal angiosarcoma.
Subsequent to diagnosis, two cycles (each cycle
lasts two weeks) of the tyrosine kinase inhibitor, sunitinib, were
administered at a dose of 37.5 mg/day. A few months later, the
patient was referred to the Gayrettepe Florence Nightingale
Hospital with recurrence of the right-sided flank pain. Subsequent
to a physical examination, abdominal magnetic resonance imaging
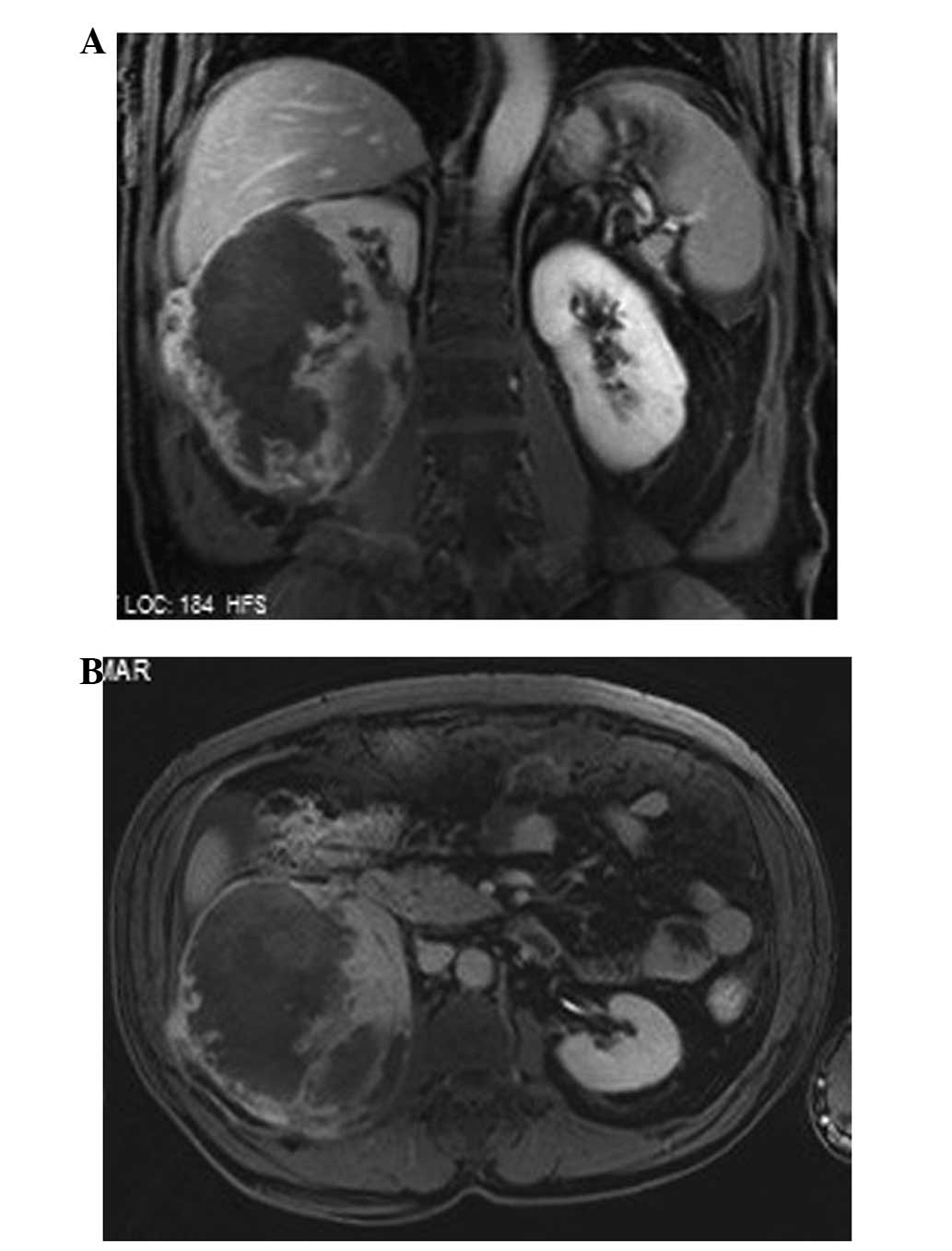
(MRI) and chest CT were performed. The abdominal MRI scan revealed
a heterogenous contrast-enhanced necrotic mass measuring 8×9.5×12.5
cm (Fig. 2) in the right
nephrectomy space, and a 6.5×11 cm pelvic metastatic mass in the
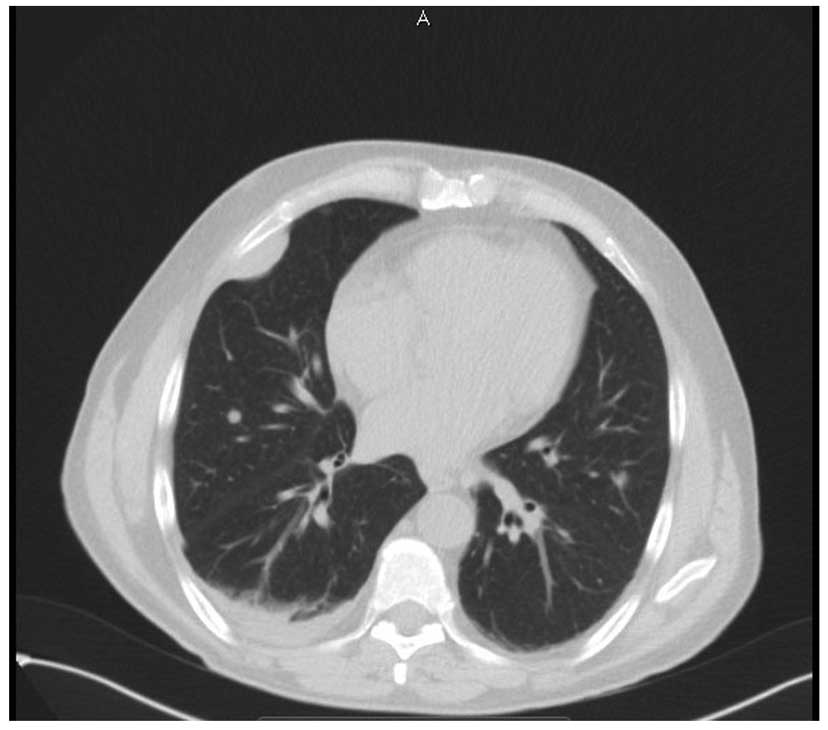
iliac fossa. CT images of the thorax identified multiple
pleuropulmonary metastatic nodules (Fig. 3). As no standard chemotherapy
protocol exists for the treatment of renal angiosarcomas, two weeks
of palliative chemotherapy was initiated with 85 mg/m2
oxaliplatin and 130 mg/m2 paclitaxel (5). In total, four cycles of chemotherapy
were administered. Despite treatment, there was no radiological
response, and only pain palliation was achieved. Following six
cycles of chemotherapy, the re-evaluation abdominal MRI and chest
CT revealed pleuropulmonary progression and no change in the size
of the right recurrent mass or pelvic mass. Due to the progressive
nature of the disease, chemotherapy with oxaliplatin and paclitaxel
was ceased. Bevacizumab (7.5 mg/kg) was then administered every
three weeks. However, there was no response to three cycles of the
antiangiogenic therapy and the patient succumbed to the disease 13
months after the initial diagnosis.
Discussion
Renal angiosarcomas are extremely rare neoplasms
that originate from the normal endothelium. Overall, the lesions
account for ~1% of all soft-tissue sarcomas (3–7). In
total, <30 cases of renal angiosarcoma have been reported since
1942, and at present, the etiology of the disease remains unknown.
The risk factors for renal angiosarcomas are uncertain, but may
include renal transplantation, post-treatment lymphedema, and
exposure to arsenic, vinyl chloride, thorotrast and radiation
(3,8,9).
Early diagnosis is challenging, as the associated
clinical symptoms do not develop in the early stages of tumor
growth, and the disease often progresses rapidly (10). Metastases are often present at the
time of diagnosis or develop due to hematogenous spread within a
couple of months or weeks after surgery. The common metastatic
sites are the lungs, liver and bones (8,11,12).
Overall, an extremely small number of patients survive more than
one year from the time of diagnosis. Clinical and radiological
findings can only identify the presence of a malignant tumor; the
definite diagnosis depends upon the histopathological examination
of the nephrectomy specimen and positive endothelial markers with
immunohistochemical staining. The most commonly used markers are
CD31, an endothelial cell adhesion molecule with high specificity
and sensitivity, and CD34, a human hematopoietic progenitor cell
antigen (3,12).
The prognosis of a patient with renal angiosarcoma
depends upon the size of the initial lesion and the presence or
absence of metastasis at diagnosis. Tumors measuring <5 cm in
diameter confer an improved prognosis compared with those >5 cm
(11,12). The five-year survival rates for
tumors >5 and <5 cm are 13 and 32%, respectively (9).
Local recurrence following surgery is common for
angiosarcomas. In the majority of cases, metastasis is present
prior to the time of diagnosis. Due to the rarity of the
malignancy, it is challenging to establish a standardized treatment
protocol for the local disease. Radical surgery with adjuvant
chemotherapy is, at present, considered to be the optimum treatment
option for angiosarcomas (9).
In conclusion, in this study the rare case of
primary renal angiosarcoma with large necrotic areas was presented,
which was firstly misdiagnosed radiologically and clinically as
necrotic renal cell carcinoma. The present case highlights the
importance of understanding the imaging features of this rare
aggressive malignancy.
References
|
1
|
DeYoung BR, Swanson PE, Argenyi ZB, et al:
CD31 immunoreactivity in mesenchymal neoplasms of the skin and
subcutis: report of 145 cases and review of putative
immunohistologic markers of endothelial differentiation. J Cutan
Pathol. 22:215–222. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prince CL: Primary angioendothelioma of
the kidney: report of a case and brief review. J Urol.
47:7871942.
|
|
3
|
Leggio L, Addolorato G, Abenavoli L, et
al: Primary renal angiosarcoma: a rare malignancy. a case report
and review of the literature. Urol Oncol. 24:307–312. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carnero López B, Fernández Pérez I,
Carrasco Alvarez JA, et al: Renal primary angiosarcoma. Clin Transl
Oncol. 9:806–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fata F, O’Reilly E, Ilson D, et al:
Paclitaxel in the treatment of patients with angiosarcoma of the
scalp or face. Cancer. 86:2034–2037. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cerilli LA, Huffman HT and Anand A:
Primary renal angiosarcoma: a case report with immunohistochemical,
ultrastructural, and cytogenetic features and review of the
literature. Arch Pathol Lab Med. 122:929–935. 1998.PubMed/NCBI
|
|
7
|
Tsuda N, Chowdhury PR, Hayashi T, et al:
Primary renal angiosarcoma: a case report and review of the
literature. Pathol Int. 47:778–783. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chaabouni A, Rebai N, Chabchoub K, Fourati
M, et al: Primary renal angiosarcoma: Case report and literature
review. Can Urol Assoc J. 7:E430–E432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Papadimitriou VD, Stamatiou KN, Takos DM,
Adamopoulos VM, et al: Angiosarcoma of kidney: a case report and
review of literature. Urol J. 6:223–225. 2009.PubMed/NCBI
|
|
10
|
Yoshida K, Ito F, Nakazawa H, Maeda Y, et
al: A case of primary renal angiosarcoma. Rare Tumors.
1:e282009.PubMed/NCBI
|
|
11
|
Akkad T, Tsankov A, Pelzer A, Peschel R,
et al: Early diagnosis and straight forward surgery of an
asymptomatic primary angiosarcoma of the kidney led to long-term
survival. Int J Urol. 13:1112–1114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sabharval S, John NT, Kumar RM and Kekre
NS: Primary renal angiosarcoma. Indian J Urol. 29:145–147. 2013.
View Article : Google Scholar
|