Introduction
Primitive neuroectodermal tumors (PNETs) are
extremely rare tumors which typically arise from multipotent
progenitor cells and are considered to be of neuroectodermal
derivation (1). Since the first
description of the condition by Hart and Earle in 1973 (2), <100 cases have been documented. The
annual incidence of this condition is estimated to range from
0.2–0.4 cases per 100,000 (3), and
its concept has been controversial for over a decade, as diagnosis
remains difficult and no effective treatment has been identified.
PNETs are rapidly growing soft tissue masses, which cause symptoms
of nerve compression and pain. They are highly malignant and
invasive, with a high rate of relapse and poor prognosis. The
five-year survival rate is 30–40% and has not altered significantly
over the last 30 years (4,5). PNET primarily occurs in children and
young adults, usually <35 years of age, with a mean age of 20
years; however, it may occur at any age, in any population and on
any limb (6).
The present study reports the case of an otherwise
healthy 60-year-old female with extradural PNET extending into the
lumbar cavity, which caused symptoms of nerve root compression. Few
such presentations have been reported previously (6–8).
Written informed consent was obtained from the patient’s
family.
Case report
A 60-year-old female was admitted to Shanghai Tenth
People’s Hospital Affiliated to Tongji University (Shanghai, China)
due to a one-year history of increasing lower back pain, worsening
numbness and pain on the back of the right thigh. The patient did
not smoke or consume alcohol, and was suffering from diabetes,
which was managed by a controlled diet. Initially, the patient’s
blood pressure was 130/80 mmHg (normal range, 100–130/60–90 mmHg),
with a regular pulse of 80 beats/min (normal range, 60–100
beats/min) and a respiratory rate of 18 breaths/min (normal range,
20–40 breaths/min). The patient was afebrile. On physical
examination, hypoesthesia was detected on the back of the thigh and
the knee jerk reflex was found to be reduced. In the straight leg
raising test an angle of 30° was achieved. The results of all
laboratory tests, including complete blood count, renal, bone,
hepatic, and coagulation profiles, lactate dehydrogenase,
carcinoembryonic antigen, α-fetoprotein, carbohydrate antigen (CA)
19-9 and CA12-5 levels, were normal. Computed tomography (CT)
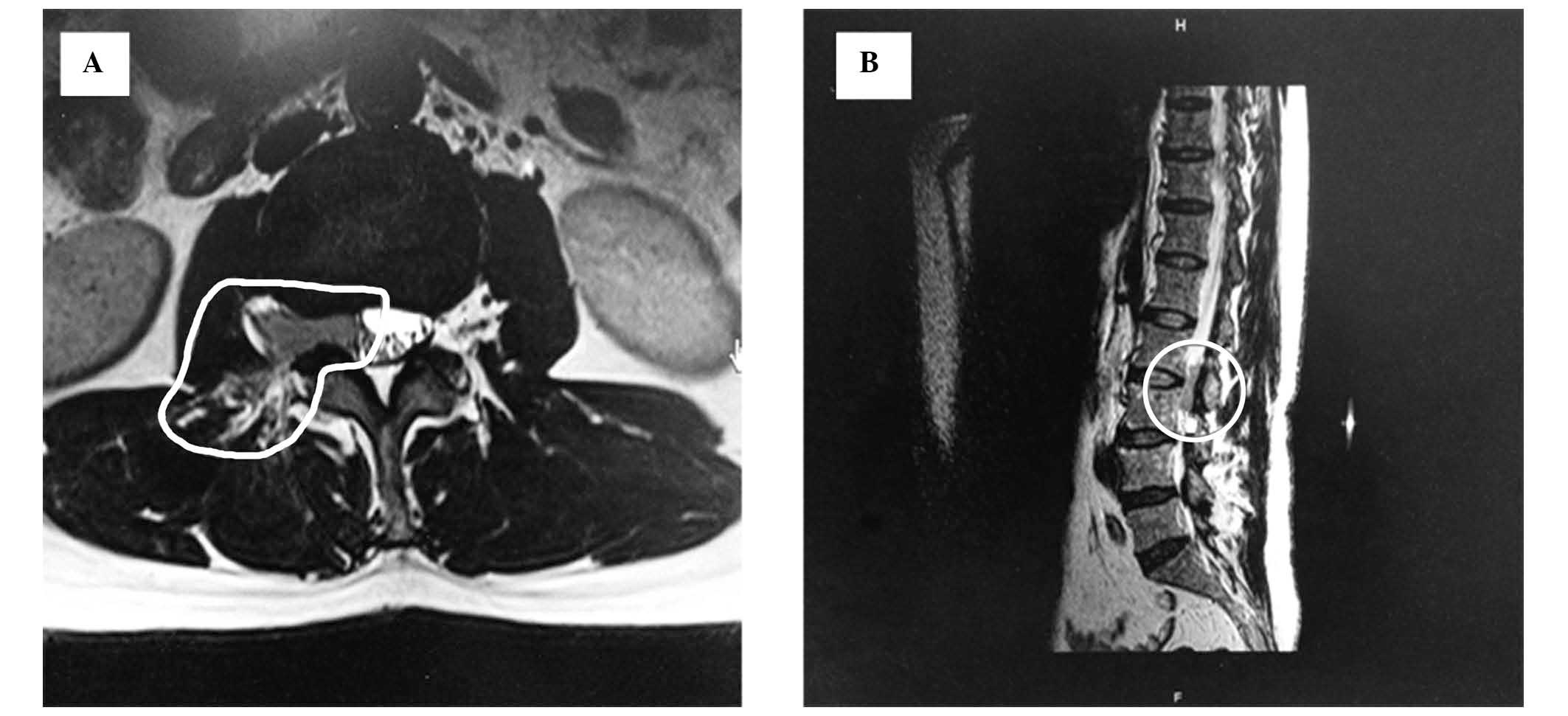
imaging of the lumbar spine revealed a 4.9×2.1×1.8 cm mass with
ill-defined margins from the L2–L3 vertebrae (Fig. 1). The lesion was isointense on the
T1-weighted image and iso- to hyperintense on the T2-weighted
image. Abdominal and pelvic CT imaging and chest X-ray did not
reveal any lesions. No evidence of ascites, or retroperitoneal or
mesenteric lymphatic metastases was observed.
The patient subsequently underwent intraspinal
neoplasm resection. Following a half laminectomy of vertebrae L2
and L3, the 4.9×2.1×1.8 cm ill-defined mass was revealed. The tumor
was adhered to the right L2 nerve root and extended into right
lumbar cavity through the dilated intervertebral foramen between L2
and L3. Following the complete removal of the mass, the L2 nerve
root was dissociated. L3 nerve root activity was improved and all
resection margins were free from tumor cells. Macroscopically, the
outer surface of the resected mass was white, irregular, firm, and
marked with several prominences, and the cut section revealed
irregular, ulcerated and necrotic tissue. Postoperatively, the
patient showed partial improvement of the deficits; lower back and
right thigh pain was alleviated and a significant improvement was
achieved in the straight leg raising test, when compared with that
observed preoperatively.
Immunohistochemical evaluation of CD99 expression
revealed characteristic reactivity on the tumor cell membranes.
Histopathological examination of the mass revealed that the tumor
cells had small round or oval morphology, strong staining for
neuron specific enolase (NSE), fine chromatin, indistinct nucleoli
and signs of mitosis. Homer-Wright rosettes were observed, composed
of tumor cells surrounding a central region of filamentous fibers
combined with necrotic blood vessel and tumor tissue. This led to a
diagnosis of PNET.
Focal radiation therapy to the spinal cord (L1–L5)
(total dose, 50.4 Gy) was administered over a period of six weeks,
however, chemotherapy was refused. Four months following surgery
and radiotherapy, the tumors recurred and the patient succumbed to
the disease four months later.
Discussion
PNETs may be classified as central (cPNET) or
peripheral (pPNET), and both types have poor prognoses (9). pPNET refers to a group of malignant
cells with similar morphological and cytological characteristics
and gene expression, that occur in the extracranial soft tissue of
the skeletal system (6). This
includes several conditions: Ewing’s sarcoma (ES), which occurs
intraosteally and extraskeletally; pPNET, which occurs in soft
tissue such as paravertebral, abdominal, pelvic or retroperitoneal
areas; and Askin tumors, which commonly arise in the chest area
(10–12). pPNET and ES are closely related
malignancies with small round cells observable on histological
examination (13). pPNETs and ES
strongly express the glycoprotein p30/32 (CD99), which is encoded
by the microneme protein 2 (MIC2) gene. CD99 is expressed in almost
all cases in a characteristic membranous manner, however, it is not
specific to this condition. The majority of PNET tumor cells stain
with vimentins, and neural markers, such as NSE, are frequently
expressed (14). PNET has also been
shown to stain with keratin in certain cases. Due to their
immunohistochemical, ultrastructural and molecular similarities,
pPNETs and ES have been categorized into the Ewing family of
tumors, which share the same chromosomal translocation but differ
in their degree of neural differentiation (15).
Primary spinal PNETs are now more frequently
diagnosed due to the improvement of histological techniques,
particularly immunohistochemistry. Initially, these cases were
diagnosed as astrocytoma, neurofibroma or ependymoma dependent upon
on the location of the tumor in relation to the dura (6). Neurofibroma was highly suspected in
the current case due to the expansion of the mass into the lumbar
cavity through intervertebral foramina; neurofibromas have
favorable prognosis if fully excised.
pPNET is a highly aggressive cancer associated with
poor prognosis. Surgical excision cannot remove the lesion
completely, leading to a postoperative recurrence rate of 90%. The
current recommended treatment for pPNET is a combined therapy
including surgery, high-dose local radiotherapy and chemotherapy
(16).
Although primary PNETs may be indicated by
radiological features, immunohistochemical evaluation following
surgery is required to confirm the diagnosis. Histological
examination must be conducted in every suspected case to diagnose
this type of tumor. The current study illustrates a unique case of
intraspinal PNET that does not match the typical age distribution
for this condition; few similar cases have been previously reported
(6–8). Radiological findings in combination
with histological examination must be used to differentiate between
PNETs and short duration paraparesis.
Despite improvements in surgical techniques and
adjuvant chemotherapy, the prognosis of this malignancy remains
poor. Further research into the molecular pathogenesis of PNET and
its potential application in immunotherapy is crucial for the
development of more effective treatments for this condition.
References
|
1
|
Rorke LB: The cerebellar medulloblastoma
and its relationship to primitive neuroectodermal tumors. J
Neuropathol Exp Neurol. 42:1–15. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hart MN and Earle KM: Primitive
neuroectodermal tumors of the brain in children. Cancer.
32:890–897. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Valle JW, Eatock M, Clueit B, et al: A
systematic review of non-surgical treatments for pancreatic
neuroendocrine tumors. Cancer Treat Rev. 40:376–389. 2014.
View Article : Google Scholar
|
|
4
|
Pape UF, Böhmig M, Berndt U, et al:
Survival and clinical outcome of patients with neuroendocrine
tumors of the gastroenteropancreatic tract in a german referral
center. Ann N Y Acad Sci. 1014:222–233. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marinsek ZP, Kavalar R and Jereb B: Ewing
sarcoma/PNET: 27 years of experience in Slovenia. Pediatr Hematol
Oncol. 23:355–367. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Patnaik A, Mishra SS, Mishra S and Deo RC:
Review of spinal neuroectodermal tumor. Br J Neurosurg. 27:2–6.
2013. View Article : Google Scholar
|
|
7
|
Virani MJ and Jain S: Primary intraspinal
primitive neuroectodermal tumor (PNET): a rare occurrence. Neurol
India. 50:75–80. 2002.PubMed/NCBI
|
|
8
|
Nayak PK, Rao KM, Sahoo GC and Mahapatra
AK: Primary thoracic primitive neuroectodermal tumor mimicking as
neurofibroma. J Neurol India. 59:648–649. 2011. View Article : Google Scholar
|
|
9
|
Kampman WA, Kros JM, De Jong TH and Lequin
MH: Primitive neuroectodermal tumours (PNETs) located in the spinal
canal; the relevance of classification as central or peripheral
PNET: case report of a primary spinal PNET occurrence with a
critical literature review. J Neurooncol. 77:65–72. 2006.
View Article : Google Scholar
|
|
10
|
Khong PL, Chan GC, Shek TW, et al: Imaging
of peripheral PNET: common and uncommon locations. Clin Radiol.
57:272–277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dehner LP: Peripheral and central
primitive neuroectodermal tumors. A nosologic concept seeking a
consensus. Arch Pathol Lab Med. 110:997–1005. 1986.PubMed/NCBI
|
|
12
|
Winer-Muram HT, Kauffman WM, Gronemeyer SA
and Jennings SG: Primitive neuroectodermal tumors of the chest wall
(Askin tumors): CT and MR findings. AJR Am J Roentgenol.
161:265–268. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hisaoka M, Hashimoto H and Murao T:
Peripheral primitive neuroectodermal tumour with
ganglioneuroma-like areas arising in the cauda equina. Virchows
Arch. 431:365–369. 1997. View Article : Google Scholar
|
|
14
|
Nutman A, Postovsky S, Zaidman I, et al:
Primary intraspinal primitive neuroectodermal tumor treated with
autologous stem cell transplantation: case report and review of the
literature. Pediatr Hematol Oncol. 24:53–61. 2007. View Article : Google Scholar
|
|
15
|
Kumar R, Reddy SJ, Wani AA and Pal L:
Primary spinal primitive neuroectodermal tumor: case series and
review of the literature. Pediatr Neurosurg. 43:1–6. 2007.
View Article : Google Scholar
|
|
16
|
Shah JP, Jelsema J, Bryant CS, et al:
Carboplatin and paclitaxel adjuvant chemotherapy in primitive
neuroectodermal tumor of the uterine corpus. Am J Obstet Gynecol.
200:e6–e9. 2009. View Article : Google Scholar
|