Introduction
Glioblastoma multiforme (GBM) is one of the most
malignant tumors of the central nervous system and exhibits a poor
prognosis, despite technological advances in surgical resection
radiotherapy and chemotherapy. The average survival time is 8–12
months, due to the high invasiveness and heterogeneity (1,2).
Temozolomide (TMZ), an orally administered alkylating agent, has
been revealed to provide significant survival benefits for patients
with GBM. Although maximal surgical resection combined with TMZ
therapy and irradiation has significantly improved the quality of
life and prolonged the survival time of patients with GBM, the
overall clinical prognosis remains unsatisfactory due to intrinsic
or acquired tumor cell resistance to TMZ (3). Previous studies have demonstrated that
O6-methylguanine-DNA methyltransferase (MGMT) is the
main factor responsible for chemoresistance to TMZ in GBM cells
(4,5).
However, the molecular mechanism of TMZ resistance is more complex
than a simple dependence on MGMT expression.
Nuclear factor-κB (NF-κB) is a component in the
major inflammatory transcription pathway that is associated with
glioblastoma and excessively activated by various cytokines and
growth factors (6). NF-κB proteins
include a family of structurally-similar transcription factors, of
which the p65/p50 heterodimer is the key form. Normally, NF-κB
exists in the cytosol and is bound to the inhibitory IκBα in an
unstimulated state. Classical signaling of the NF-κB activation
pathway consists of one of several extracellular ligands, such as
TNF-α, IL-1 or LPS, binding to the NF-κB receptor, which in turn
activates the IκB kinases (IKKs) IKKα and IKKβ, the latter being
particularly important as it phosphorylates IκBα and enables IκBα
ubiquitination and degradation, thus releasing and activating the
NF-κB protein. Activated NF-κB translocates to the nucleus and
binds to DNA, which initiates the transcription of several genes
(7,8).
Excessive and aberrant activation of NF-κB has been
identified in GBM, and as the oncogene in glioma, the levels of
NF-κB activity are much higher in GBM tissue compared with non-GBM
tissue, and has also been associated with a worse prognosis in GBM
(9). In addition, NF-κB has been
reported to be associated with TMZ resistance primarily due to the
anti-apoptotic activity of NF-κB (10). Therefore, the present study proposed a
hypothesis that down-regulation of NF-κB via the IκBα inhibitor
reversed the resistance to TMZ, and detected the link between the
NF-κB pathway and MGMT expression in a TMZ resistant (TR) cell
line.
Materials and methods
Drugs and reagents
TMZ was purchased from Tasly Pharmaceutical Co.,
Ltd. (Tianjin, China). The stock solutions were prepared in
dimethyl sulphoxide (DMSO) at a concentration of 10 mM, and the
solutions were diluted in the cell culture medium to the respective
final concentrations. Sulforhodamine B (SRB) was obtained from
Sigma-Aldrich (St. Louis, MO, USA). The IκBα inhibitor BAY 11-7082
was purchased from Beyotime Institute of Biotechnology (Haimen,
Jiangsu, China). All antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA), consisting of primary mouse
anti-human polyclonal antibodies against MGMT (cat. no.
sc-400720-HDR), NF-κB (p65; cat. no. sc-8008 AC) and β-actin (cat.
no. sc-77326-PR), rabbit anti-human polyclonal antibodies against
phosphorylated p65 (cat. no. sc-45101), B-cell lymphoma (Bcl)-2
(cat. no. sc-130916) and Bcl-2-associated X protein (cat. no.
sc-377576), and secondary HRP-conjugated rabbit anti-goat (cat. no.
sc-2768) antibodies.
Cell lines and culture conditions
The human malignant glioma U251 cell line, obtained
from the Institute of Biochemistry and Cell Biology (Shanghai
Institutes for Biological Sciences, Chinese Academy of Sciences,
Shanghai, China), was cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% heat-inactivated fetal bovine
serum (FBS), 100 units/ml of penicillin and 100 µg/ml of
streptomycin in a humidified atmosphere containing 5%
CO2 at 37°C. All media and sera were purchased from
Gibco Life Technologies (Carlsbad, CA, USA).
To establish the TR cell line, the U251 cells were
initially exposed to 1.25 µM TMZ for two weeks, and the
concentration of TMZ was then doubled for 4–8 weeks until it
reached 160 µM. After ~10 months, the resistant clones were
isolated and confirmed using an SRB assay. The TR variants
generated by this method were denoted as TR/U251 cells. The
parental U251 cells and the cells treated with DMSO alone were
included for parallel analysis.
Chemosensitivity assay
For the chemosensitivity assay, 2,000 U251 or
TR/U251 cells were plated in each well of a 96-well plate, at 37°C
in a 5% CO2 atmosphere. After 24 h, the culture media
were replaced with fresh media in the absence or presence of serial
concentrations of TMZ. After 96 h, the SRB assay was used to
determine the surviving cell numbers using optical density (OD), as
measured by absorbance at 490 nm using an ELISA reader (Multiskan
MK3; Thermo Fisher Scientific, Waltham, MA, USA). The percentage of
cell viability was calculated relative to the untreated cells,
which acted as the control. A dose-response curve was plotted, and
the 50% inhibiting concentration (IC50) values for TMZ
were calculated by the derivation of the best-fit lines using three
independent experiments, which were performed in triplicate.
Cell proliferation assay
In total, 5,000 cells/well were seeded in 96-well
plates. The cells were serum-starved for 24 h and were treated
again with serum in medium prior to the start of the experiment.
The relative density of cells was determined using the SRB assay on
a daily basis over a 72-h period. Absorbance was measured at 490 nm
in three independent experiments performed in triplicate.
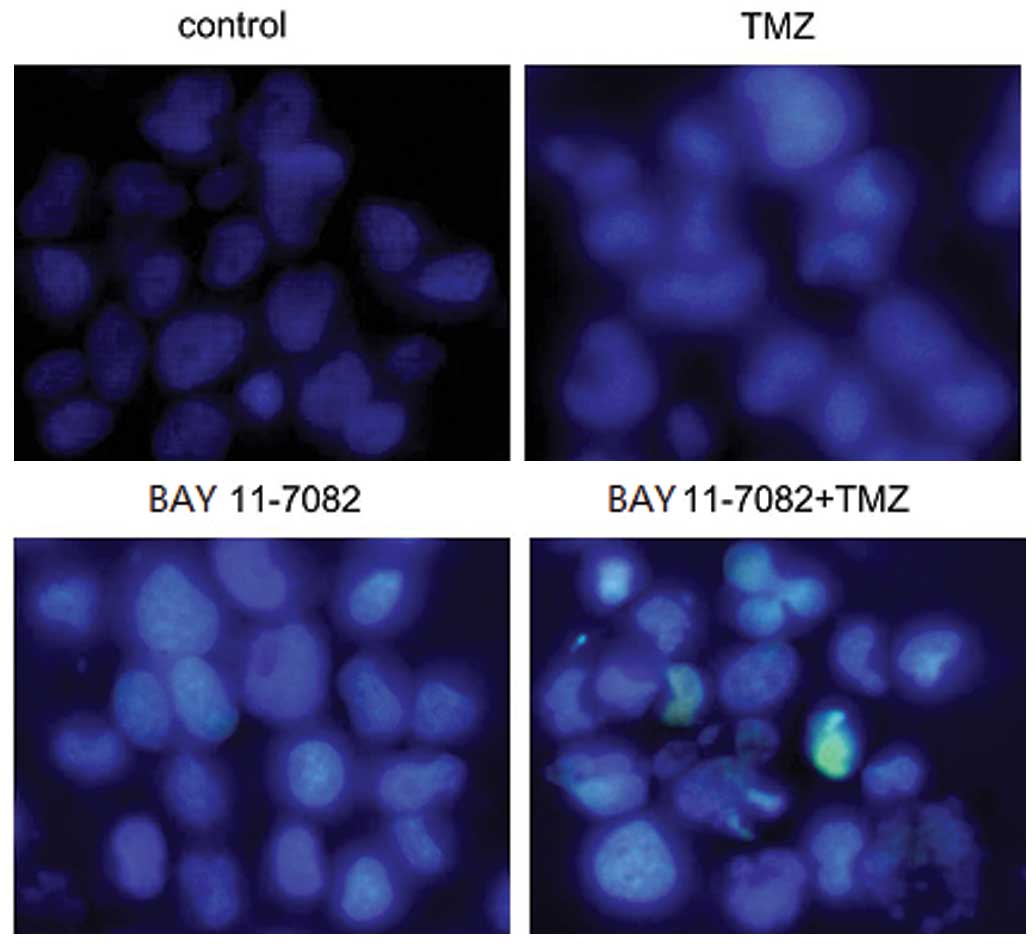
Hoechst 33258 nuclear staining
For nuclear staining, ~1×105 cells/well
were seeded in six-well plates, and treated with TMZ or BAY
11-7082, respectively. The cells were washed with 0.01 M PBS, fixed
with 70% ethanol for 2 h at 4°C, and stained with 5 µg/ml Hoechst
33258. The change in the cell nuclei was then observed using a
fluorescence microscope at a magnification of x200 (Olympus, Tokyo,
Japan).
Western blot analysis
Western blot analysis was performed using standard
techniques, as follows. Briefly, the cells in the culture flasks
were lysed in mammalian protein extraction reagent (Thermo Fisher
Scientific, Waltham, MA, USA), and the whole protein concentrations
were determined using the bicinchoninic acid protein assay kit from
Pierce Biotechnology, Inc. (Rockford, IL, USA). The protein was
then separated by 10% SDS-PAGE and transferred to polyvinylidene
fluoride membranes (EMD Millipore; Billerica, MA, USA). The
membranes were blocked using 5% non-fat dry milk in Tris-buffered
saline with Tween 20 (TBST; Guidechem, Shanghai, China) for 1 h and
then incubated with primary antibodies overnight at 4°C. Subsequent
to three washes with TBST, the membranes were incubated three times
with secondary antibodies conjugated to horseradish peroxidase
(1:5,000) for 2 h at room temperature. Finally, the
immunoreactivity was detected using an enhanced chemiluminescence
procedure kit (Thermo Fisher Scientific) and accessed using a
quantitative gel and western blotting imaging system
(Becton-Dickinson, Franklin Lakes, NJ, USA). The protein expression
was quantified subsequent to normalization to β-actin.
Statistical analysis
Each study was replicated in at least three
independent experiments. SPSS 13.0 statistical software (SPSS,
Inc., Chicago, IL, USA) was used to perform the statistical
analysis. The data are presented as the mean ± standard deviation.
One-way analysis of variance (ANOVA) and Student's t-test
were used to analyze the significance of the differences between
study groups. The least significant difference method for multiple
comparisons was used to compare the parental and control groups
when ANOVA indicated a statistical significance between the groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Generation of the TR/U251 cell
line
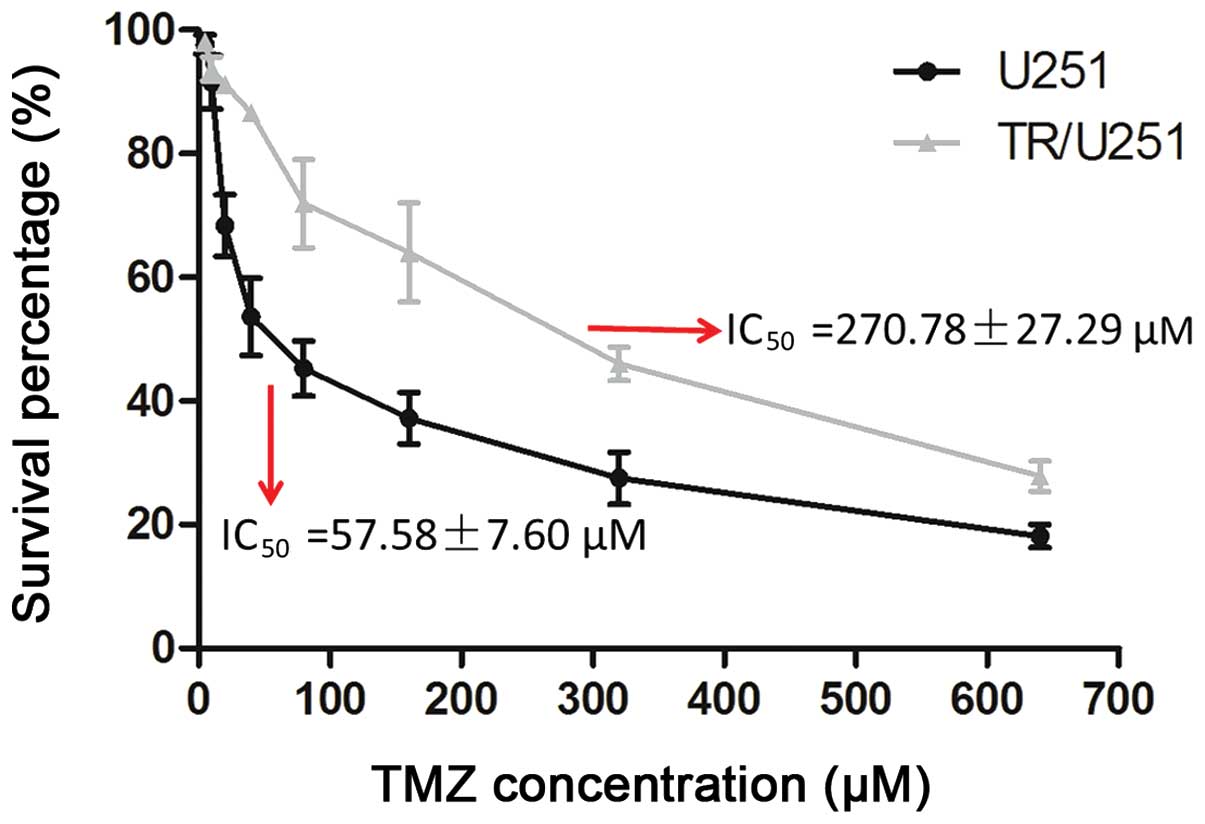
The TR/U251 cell line was generated from the
parental U251 cell line by initially exposing the cells to 1.25 µM
TMZ for two weeks, and then doubling the TMZ concentration every
two passages over a period of 4–8 weeks, with the doses ranging
between 12.5 and 160 µM. The IC50 of TMZ in the TR/U251
cells was 270.78±27.29 µM and the IC50 of TMZ in the
parental U251 cells was 57.58±7.60 µM. Therefore, the TR/U251 cells
were approximately five times more resistant to TMZ compared with
the parental cells (Fig. 1). The TMZ
resistance of the TR/U251 cells was maintained with the absence of
drugs for approximately eight weeks. In addition, the TR cells
exhibited constant resistance subsequent to TMZ withdrawal, which
was maintained for an almost three-month period of time (data not
shown).
High expression of MGMT and NF-κB in
TR/U251 cells compared with the parental cells
MGMT, the DNA repair protein, contributed to drug
resistance through the demethylation of the methyl added to
O6-methylguanine by TMZ (11). Additionally, the inflammatory
transcription factor NF-κB has been demonstrated to be highly
expressed and activated in the GBM cells and can be induced by
anticancer drugs, including the alkylating agent TMZ (12,13). To
investigate whether the TMZ resistance that occurred in TR cells
was due to the expression of MGMT or NF-κB, the expression levels
of MGMT, NF-κB and phosphorylated NF-κB were investigated by
western blot analysis of the parental and TR/U251 cells. Western
blot analysis detected high MGMT expression in the TR/U251 cells,
but an extremely low or hardly-detected expression in the parental
U251 cells. Similarly, the levels of phosphorylated-P65, a subunit
of NF-κB, were increased in the TR/U251 cells compared with the
parental cells. No evident difference in NF-κB (P65) expression was
found between the two cell lines. The TR/U251 cells exhibited an
~5.4-fold increase in the level of phosphorylated-P65 (Fig. 2). Accordingly, the results indicated
that TMZ resistance acquired in TR cells may be associated with
MGMT or NF-κB.
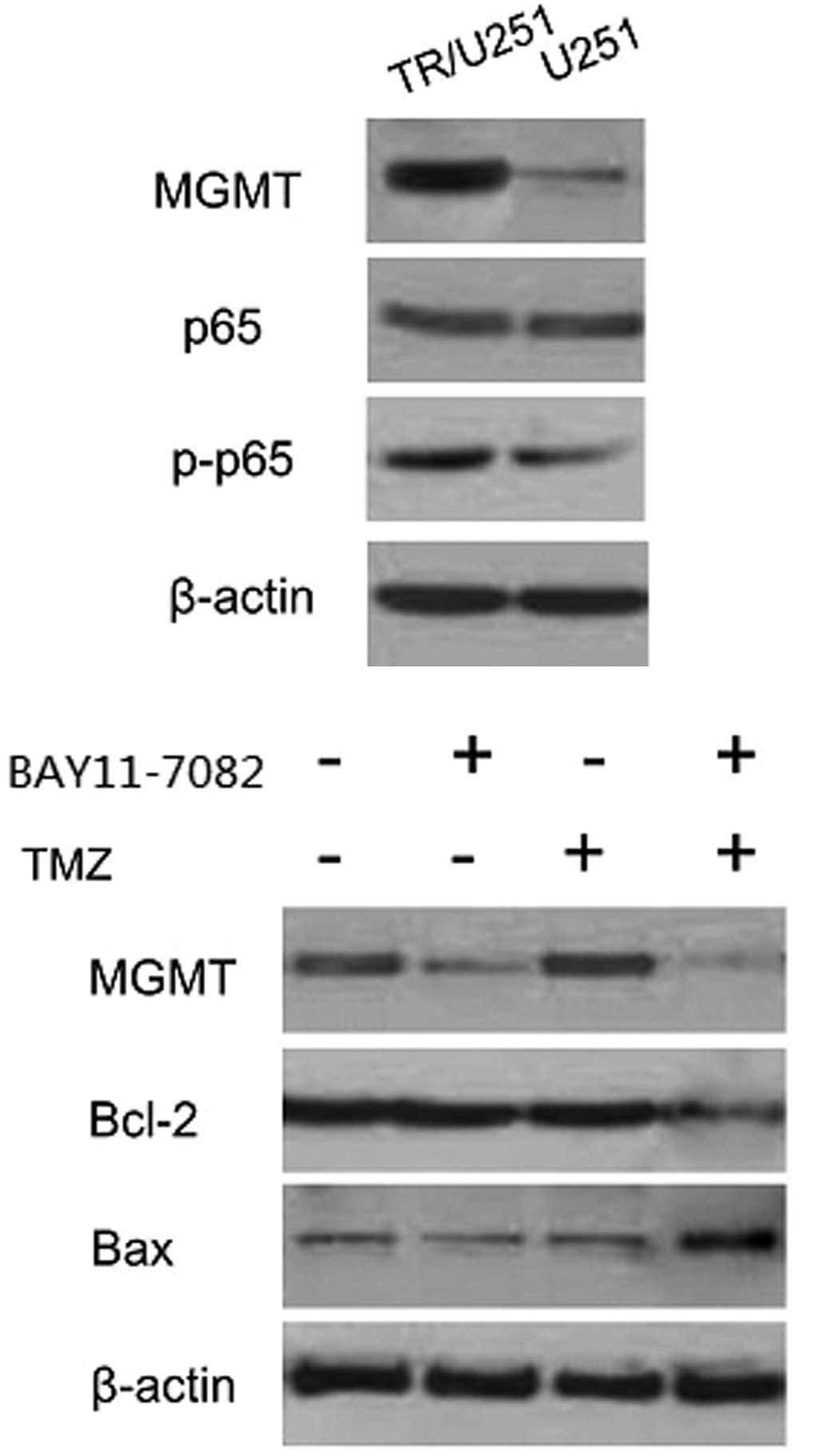 | Figure 2.Expression of MGMT, Bcl-2 and p65 in
the BAY 11-7082, TMZ and combination treatment groups. A high
expression of MGMT was found in the TR/U251 cells, while expression
was hardly detected in the U251 cells. Similarly, the levels of
phosphorylated-P65 were increased in the TR/U251 cells compared
with the U251 cells. No statistical difference was found in the
NF-κB expression, assessed through the detection of the p65
subunit, between the two cell types. The TR/U251 cells demonstrated
a ~5.4-fold higher level of phosphorylated-P65 compared with the
U251 cells. MGMT was hardly detected when cells were cultured with
a combination of TMZ and BAY 11-7082. A higher level of MGMT was
detected in the cells treated with TMZ or BAY 11-7082 separately.
The expression of Bcl-2 and Bax was reversed only in the cells
treated with a combination of TMZ and BAY 11-7082. TMZ,
temozolomide; TR, TMZ-resistant; MGMT,
O6-methylguanine-DNA methyltransferase; Bcl-2, B-cell
lymphoma-2; Bax, Bcl-2-associated X protein; p-p65, phosphorylated
p65. |
BAY 11-7082 enhances TMZ-induced
cytotoxicity in TR/U251 cells
Since a high expression of NF-κB was detected in the
TR/U251 cells compared with the parental cells, the IκBα inhibitor
BAY 11-7082 was used to determine whether inhibition of NF-κB
sensitizes TR/U251 cells to TMZ-induced cell death. The
morphological and cell proliferation assays for TMZ were performed
in the presence and absence of BAY 11-7082. To evaluate the
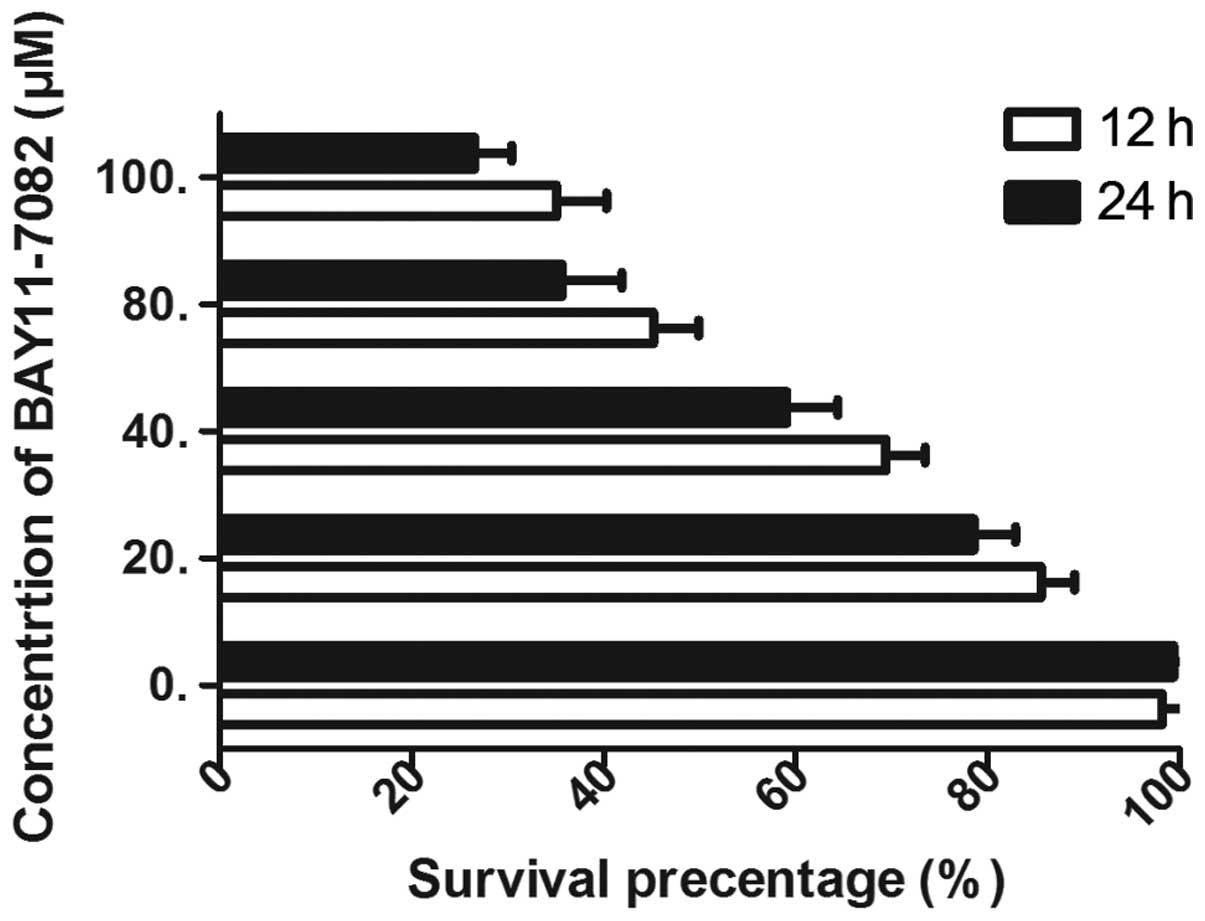
inhibition efficiency of BAY 11-7082, the TR/U251 cells were
treated with various concentrations of BAY 11-7082. The cell
survival rate was found to be inhibited in a dose- and
time-dependent manner by BAY 11-7082 and the efficacy was maximized
at 100 µM (Fig. 3). Therefore, the
IC50 of 50 µM was selected to combine with TMZ. In
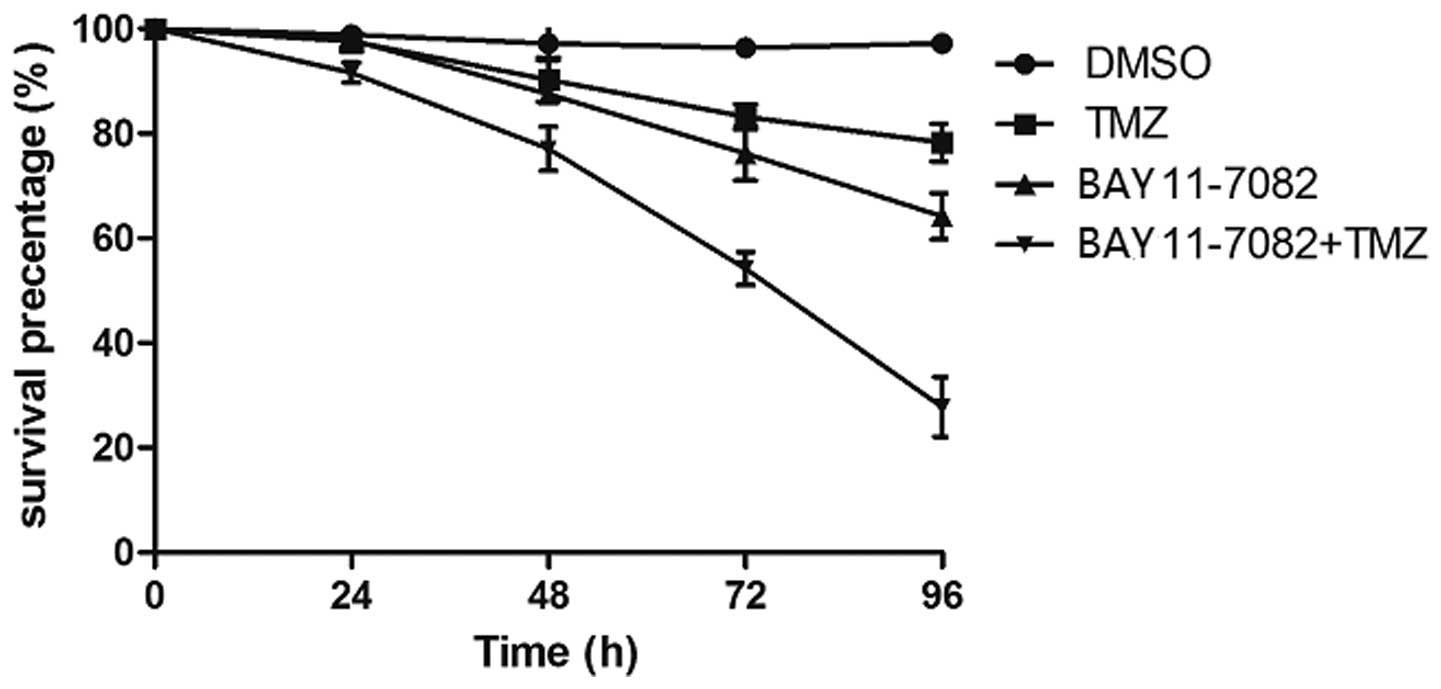
comparison to treatment with TMZ or BAY 11-7082 alone, there was
significant damage to the morphology of cells subsequent to
treatment with TMZ in combination with BAY 11-7082. When exposed to
80 µM TMZ for 24–96 h, the survival rate for the TR/U251cells
slowly decreased between 97.6±1.4 and 78.3±3.6%, while exposure to
50 µM BAY 11-7082 decreased the cell viability between 97.9±2.3 and
64.2±4.4%. By contrast, the survival rate markedly decreased
between 91.6±1.9 and 27.8±5.7% when the cells were exposed to 80 µM
TMZ in combination with 50 µM BAY 11-7082 (P<0.01; Fig. 4). These results revealed that
combination treatment with BAY 11-7082 and TMZ resulted in
synergistic decreases in cell viability as compared to each
treatment alone.
BAY 11-7082 suppresses the expression
of MGMT and enhances the TMZ-induced apoptosis in TR/U251
cells
To further explore the molecular mechanism
underlying the effect of BAY 11-7082 on TR cells, an experiment was
performed to assess the hypothesis that inhibition of proliferation
may be due to changes in the expression of MGMT and pro-apoptotic
proteins. First, the expression of NF-κB was detected at various
concentrations of BAY 11-7082 for 24 h in TR/U251 cells. As
Fig. 4 shows, the optimal
concentration of BAY 11-7082 was 20 µM and the optimal
concentration of TMZ was 100 µM, which was one-third of the
IC50 of TMZ in the TR/U251 cells. Based on these
conditions, the TR/U251 cells were incubated with 100 µM TMZ or 20
µM BAY 11-7082 alone, or co-treated with BAY 11-7082 and TMZ.
Western blot analysis revealed that exposure to BAY 11-7082 alone
significantly decreased the level of MGMT in TR/U251 cells compared
with the DMSO treatment. However, combination treatment with TMZ
completely suppressed the level of MGMT. In addition, the
expression of Bcl-2 was elevated following incubation with BAY
11-7082, alone and in combination with TMZ, whereas the decreased
level of caspase 3 was observed following treatment with BAY
11-7082 alone or in combination with TMZ (Fig. 5). Therefore, this result was
consistent with the association between NF-κB and TMZ resistance,
indicating that the inhibition of NF-κB is able to reverse TMZ
resistance in TR cells.
Discussion
TMZ chemotherapy combined with surgery and
radiotherapy is currently regarded as the standard treatment in
glioma. Although oral administration of TMZ is the most effective
strategy for the chemotherapy of glioma, its clinical efficacy is
limited in the majority of cases by the recurrence or development
of intrinsic or acquired TMZ resistance (14). Since the current understanding of
mechanism behind TMZ resistance remains unclear, the present study
aimed to establish a resistant cell line model to explore the
molecular mechanism of TMZ resistance in glioma.
As a DNA repair enzyme, MGMT is a protein that can
transfer the alkylated group of methylguanine from the
O6 position to its own cysteine residue (4,5,15), so that the TMZ-induced guanine damage
in DNA can be repaired. Previous studies have demonstrated that the
levels of MGMT vary considerably in glioma cells, which can be
divided into the MGMT repair-deficient cells and cells with a
sufficient MGMT repair (16).
Provided that the cytotoxicity effects of the alkylating agent can
be reduced in cells with a high level of endogenous MGMT activity,
the MGMT expression may be considered to be the most important
molecular predictor of TMZ resistance and prognosis in glioma. In
the present study, a TR glioma cell model was successfully
generated by exposure of parental U251 cells to a gradually
increasing TMZ concentration. The resulting TR/U251 cells exhibited
a strong resistance to TMZ, demonstrating an IC50 that
was six times higher than the IC50 of the parental
cells, and also demonstrated a good stability for almost three
months subsequent to TMZ withdrawal. Notably, the TR/U251 cell line
exhibited high levels of MGMT and also demonstrated a more stable
morphology and more vigorous cell proliferation compared with the
parental U251 cells. Accordingly, it was hypothesized that this may
be associated with the high expression of MGMT in the TR/U251
cells, as MGMT-positive cells selectively survive during the
stepwise screening process. However, MGMT-negative cells are
eliminated, which is in agreement with the earlier stepwise
inducing method that established the anticancer drug-resistant
cells.
Constitutively activated NF-κB is one of the major
transcription factors associated with GBM and it may be responsible
for aspects of cancer development that include the inhibition of
apoptosis, invasion, immune response and inflammation (9). Previous studies have demonstrated that
GBM tissues possess constitutive NF-κB activation, which is also
associated with the grade of GBM and a worse prognosis (10,11).
Additionally, increasing evidence demonstrates the key role of the
NF-κB signaling pathway in the drug resistance of GBM. NF-κB is
activated in response to treatment with cytotoxic drugs, such as
taxanes, vinca alkaloids and topoisomerase inhibitors. Similarly,
alkylating agents, including carmustine and TMZ, can also activate
NF-κB through DNA damage pathway activation (10,17). In
the present study, stepwise exposure to TMZ was found to result in
a marked increase in the NF-κB subunit P65 and phosphorylated-P65
compared with the parental cell. Inactivated NF-κB usually locates
to the cytoplasm. Upon DNA damage caused by alkylating agents,
activated ataxia telangiectasia mutated kinase triggers multiple
events to promote cell survival and facilitate repair, including
the activation of NF-κB (18).
Therefore, the vigorous cell proliferation may also be due to the
constant self-repair of DNA damage and NF-κB activation in GBM.
Taking into account that MGMT and NF-κB are each
highly expressed in the TR/U251 cells, then the association between
the two may be associated with TMZ resistance. The present study
revealed that the IκBα inhibitor BAY 11-7082 combined with TMZ
markedly suppressed the level of MGMT in TR/U251 cells and promoted
the initiation of TMZ-induced apoptosis (Fig. 2), indicating the key role of NF-κB in
the regulation of MGMT expression. Previous studies have also
demonstrated that forced expression of the NF-κB p65 subunit in
HEK293 cells induced an increase in MGMT expression, whereas a lack
of NF-κB completely abrogated the induction of apoptosis,
independent of MGMT promoter methylation (19). Furthermore, resveratrol reverses TMZ
resistance by regulation of MGMT in T98 GBM cells in a
NF-κB-independent manner (20). The
aforementioned studies revealed a significant correlation between
the extent of NF-κB activation and MGMT expression in glioma cells.
Provided that NF-κB and MGMT are each involved in the regulation of
TMZ resistance, the two can also be considered to be promising
targets for novel therapeutic approaches in glioma. Therefore, the
inhibition of NF-κB can decrease MGMT activity and combine with TMZ
treatment to possibly offer clinical benefits for glioma patients
exhibiting TMZ resistance.
References
|
1
|
Tate MC and Aghi MK: Biology of
angiogenesis and invasion in glioma. Neurotherapeutics. 6:447–457.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grauer OM, Wesseling P and Adema GJ:
Immunotherapy of diffuse gliomas: biological background, current
status and future developments. Brain Pathol. 19:674–693. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mason WP: Emerging drugs for malignant
glioma. Exprt Opin Emerg Drugs. 13:81–94. 2008. View Article : Google Scholar
|
|
4
|
Auger N, Thillet J, Wanherdrick K, et al:
Genetic alterations associated with acquired temozolomide
resistance in SNB-19, a human glioma cell line. Mol Cancer Ther.
5:2182–2192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hegi ME, Diserens AC, Godard S, et al:
Clinical trial substantiates the predictive value of
O-6-methylguanine-DNA methyltransferase promoter methylation in
glioblastoma patients treated with temozolomide. Clin Cancer Res.
10:1871–1874. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar A, Takada Y, Boriek AM, et al:
Nuclear factor-kappaB: its role in health and disease. J Mol Med
(Berl). 82:434–448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Senftleben U, Cao Y, Xiao G, et al:
Activation by IKKalpha of a second, evolutionary conserved,
NF-kappaB signaling pathway. Science. 293:1495–1499. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Naugler WE and Karin M: NF-kappaB and
cancer-identifying targets and mechanisms. Curr Opin Genet Dev.
18:19–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kapoor GS, Zhan Y, Johnson GR and O'Rourke
DM: Distinct domains in the SHP-2 phosphatase differentially
regulate epidermal growth factor receptor/NF-kappaB activation
through Gab1 in glioblastoma cells. Mol Cell Biol. 24:823–836.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bredel M, Bredel C, Juric D, et al: Tumor
necrosis factor-alpha-induced protein 3 as a putative regulator of
nuclear factor-kappaB-mediated resistance to O6-alkylating agents
in human glioblastomas. J Clin Oncol. 24:274–287. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crinière E, Kaloshi G, Laigle-Donadey F,
et al: MGMT prognostic impact on glioblastoma is dependent on
therapeutic modalities. J Neuro-oncol. 83:173–179. 2007. View Article : Google Scholar
|
|
12
|
Bottero V, Busuttil V, Loubat A, et al:
Activation of nuclear factor kappaB through the IKK complex by the
topoisomerase poisons SN38 and doxorubicin A brake to apoptosis in
HeLa human carcinoma cells. Cancer Res. 61:7785–7791.
2001.PubMed/NCBI
|
|
13
|
Cusack JC Jr, Liu R and Baldwin AS Jr:
Inducible chemoresistance to 7-ethyl-10
[4-(1-piperidino)-1-piperidino] carbonyloxycamptothecin (CPT-11) in
colorectal cancer cells and a xenograft model is overcome by
inhibition of nuclear factor-κB Activation. Cancer Res.
60:2323–2330. 2000.PubMed/NCBI
|
|
14
|
Ma J, Murphy M, O'Dwyer PJ, et al:
Biochemical changes associated with a multidrug-resistant phenotype
of a human glioma cell line with temozolomide-acquired resistance.
Biochem Pharmacol. 63:1219–1228. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martinez R, Schackert G, Yaya-Tur R, et
al: Frequent hypermethylation of the DNA repair gene MGMT in
long-term survivors of glioblastoma multiforme. J Neuro-oncol.
83:91–93. 2007. View Article : Google Scholar
|
|
16
|
Rodriguez FJ, Thibodeau SN, Jenkins RB, et
al: MGMT immunohistochemical expression and promoter methylation in
human glioblastoma. Appl Immunohistochem Mol Morphol. 16:59–65.
2008.PubMed/NCBI
|
|
17
|
Kasuga C, Ebata T, Kayagaki N, et al:
Sensitization of human glioblastomas to tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL) by NF-kappaB
inhibitors. Cancer Sci. 95:840–844. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nogueira L, Ruiz-Ontañon P,
Vazquez-Barquero A, et al: The NFκB pathway: a therapeutic target
in glioblastoma. Oncotarget. 2:646–653. 2011.PubMed/NCBI
|
|
19
|
Lavon I, Fuchs D, Zrihan D, et al: Novel
mechanism whereby nuclear factor κB mediates DNA damage repair
through regulation of O(6)-methylguanine-DNA-methyltransferase.
Cancer Res. 67:8952–8959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang H, Lin H, Zhang X, et al:
Resveratrol reverses temozolomide resistance by downregulation of
MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway.
Oncol Rep. 27:2050–2056. 2012.PubMed/NCBI
|