Introduction
All types of cancer cell are prone to resisting
apoptotic cell death; therefore, knowledge of the mechanisms of
resistance to apoptosis may lead to the development of novel
therapeutic strategies (1). Changes
in the apoptotic susceptibility of cancer cells may contribute to
enhanced resistance to conventional anticancer therapies, such as
radiation and cytotoxic agents, as well as to neoplastic
development (2). Altered expression
of B-cell lymphoma-2 (Bcl-2) family members has been proposed as a
possible mechanism for the development of resistance to certain
cytotoxic antineoplastic agents. The Bcl-2 family of proteins is
composed of pro- and anti-apoptotic members that regulate cellular
proliferation and death, respectively, via inter- and intra-family
interactions (3,4). In particular, Bcl-2 has emerged as an
important clinical prognostic marker in breast cancer (5,6), being
overexpressed in ~75% of cases of breast cancer (approaching 85%
for estrogen-positive breast cancer) (6). ABT263 (navitoclax) is a small molecule
that binds with high affinity to Bcl-2 and Bcl-extra large
(Bcl-xL), and antagonizes their anti-apoptotic functions, thereby
inducing apoptosis in various cancer cell types (7,8). However,
this molecule is limited by its lack of efficacy, and its
significant toxicity and resistance (9).
Ribosomal S6 kinase 1 (S6K1) is the downstream
effector of mammalian target of rapamycin complex 1 (mTORC1), a
protein complex that regulates essential cellular functions,
including cell survival, proliferation, metabolism, migration and
angiogenesis (10). The S6K1 gene is
amplified in numerous breast cancer cell lines (11–13), and
S6K1 gene amplification or protein expression has been associated
with a poor prognosis in breast cancer (14). Thus, the targeting of S6K1 in
combination with ABT263 may present an effective strategy for the
treatment of patients with breast cancer.
Therefore, the present study aimed to examine the
combinatorial apoptotic effects of ABT263 (a Bcl-2/Bcl-xL
inhibitor) and PF4708671 (an S6K1 inhibitor) treatment on BT474
breast cancer cells.
Materials and methods
Cell culture and reagents
The breast cancer BT474 and MCF7 cell lines were
obtained from the American Type Culture Collection (Manassas, VA,
USA) and cultured in RPMI-1640 medium and Dulbecco's modified
Eagle's medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
containing 10% fetal bovine serum (Invitrogen Life Technologies).
PF4708671 was purchased from Sigma-Aldrich (St. Louis, MO, USA),
ABT263 (navitoclax) from Selleckchem (Houston, TX, USA) and
Sorafenib from Bayer Healthcare Pharmaceuticals (New Haven, CT,
USA).
Assessment of cell death
The BT474 cells were treated with 0, 0.5, 1 and 2 µM
ABT263, or 0, 5, 10 and 20 µM PF4708671. The treated cells were
stained with Annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI; BD Biosciences, San Jose, CA, USA) for the
assessment of apoptotic cell death, as previously described
(15). As a positive control, MCF7
cells were treated with 5 µM sorafenib and deprived of glucose for
24 h.
Western blot analysis
The cells were lysed in lysis buffer [50 mM Tris-HCl
(pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1%
SDS] supplemented with a protease inhibitor cocktail (Roche
Diagnostics GmbH, Mannheim, Germany). The protein concentrations
were then measured using the Bradford method (16). The cell lysates were separated via
SDS-PAGE and transferred to nitrocellulose membranes, followed by
immunoblotting with specific primary and horseradish
peroxidase-conjugated secondary antibodies. Antibodies specific for
cleaved poly(ADP-ribose) polymerase (PARP; rabbit polyclonal;
1:1,000; cat no. 9541), p-S6 at Ser240/244 (rabbit monoclonal;
1:2,000; cat no. 4838), S6K1 (rabbit polyclonal; cat no. 9202;
1:1,000) and survivin (rabbit monoclonal; 1:1,000; cat no. 2808)
were obtained from Cell Signaling Technology (Beverly, MA, USA), an
antibody for Bcl-xL (mouse monoclonal; 1:1,000; cat no. 610746) was
obtained from BD Biosciences (Franklin Lakes, NJ, USA), and an
antibody for β-actin (mouse monoclonal; 1:5,000; cat no. A5316) was
obtained from Sigma-Aldrich. The immunoreactive bands were
visualized using SuperSignal West Pico Chemiluminescent Substrates
(Thermo Scientific Pierce, Rockford, IL, USA).
Small interfering (siRNA) and
transfection
Bcl-2 (cat no. sc-29214), Bcl-xL (cat no. sc-43630)
and control (cat no. sc-37007) siRNAs were purchased from Santa
Cruz Biotechnology, Inc. (San Jose, CA, USA). S6K1 (cat no.
SI00301721) and control siRNAs (cat no. 1022076) were purchased
from Qiagen, Inc. (Valencia, CA, USA). Transient 24-h siRNA
transfections were performed with Lipofectamine® RNAiMAX reagent,
according to the manufacturer's instructions (Invitrogen Life
Technologies). The BT474 cells were transiently transfected with
the indicated siRNAs for 24 h.
Measurement of cell viability
The cell viability was determined by measuring the
mitochondrial conversion of
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide to a
colored product, as previously described (15). Images of cell populations were
captured microscopically with an Olympus CKX41 microscope (Olympus
Corporation, Tokyo, Japan).
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR) analysis
RNA isolation and RT-PCR analysis were conducted as
previously described (17). The
following primers were used: Survivin, sense,
5′-GGACCACCGCATCTCTAC-3′ and anti-sense, 5′-CAGCCTTCCAGCTCCTTG-3′,
with a 156-bp product (15); and
β-actin, sense, 5′-GGATTCCTATGTGGGCGACAG-3′ and anti-sense,
5′-CGCTCGGTGAGGATCTTCATG-3′, with a 438-bp product (15).
Isobologram analysis
The effects of two-drug combinations were assessed
by isobologram analysis. The cells were treated with different
concentrations of ABT263 (0–20 µM) and PF4708671 (0–40 µM).
Combinations resulting in 25±1% cytotoxicity were expressed as a
percentage of each single drug, alone, producing an equivalent
level of cytotoxicity [fractional inhibitory concentrations
(FIC)=concentration of each drug in the combination/concentration
of each drug alone]. When the sum of the FIC was 1, the combination
was additive and the graph was expressed as a straight line; when
the sum was <1, the combination was synergistic and the graph
demonstrated a concave shape; and when the combination was >1,
the combination was antagonistic and the graph demonstrated a
convex shape.
Statistical analysis
Data are expressed as the mean ± standard deviation
of three independent experiments. A Student's t-test was performed
to analyze differences between groups. All statistical analyses was
performed using SigmaPlot software (version 11; Systat Software
Inc., San Jose, CA, USA) and P<0.05 was considered to indicate a
statistically significant difference.
Results
Individual treatment with ABT263 or
PF4708671 has no effect on cell death
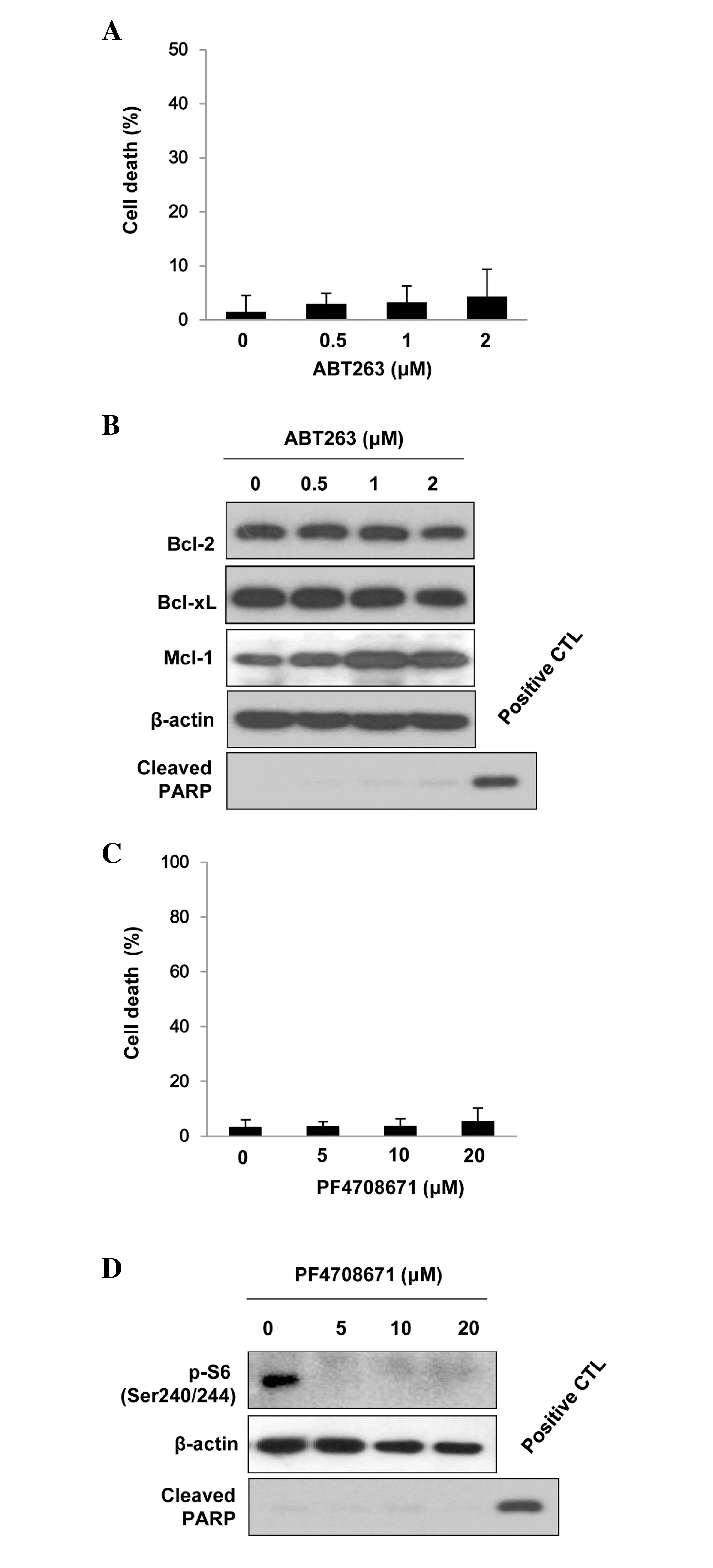
Initially, the effects of ABT263 on BT474 on breast
cancer cell apoptosis were examined. Apoptosis assays were
performed using Annexin V/PI staining and western blot analysis of
cleaved PARP expression. As indicated in Fig. 1A and B, treatment with ≤2 µM ABT263
did not induce marked apoptotic cell death in the BT474 cells.
Furthermore, protein expression levels of myeloid cell leukemia-1
[Mcl-1 (a Bcl-2-related protein)] were upregulated upon treatment
with ABT-263, however, the expression levels of Bcl-2 and Bcl-xL
did not change (Fig. 1B).
BT474 breast cancer cells are characterized by the
overexpression of S6K1 (18).
PF4708671 treatment suppressed S6K1 activity in the BT474 cells, as
evidenced by the decreased phosphorylation of S6 at Ser240/244
(Fig. 1C). However, PF4708671 (≤20
µM) resulted in no significant cell death (Fig. 1D). These data indicate that ABT263 or
PF4708671 treatment alone is not sufficient to cause apoptosis.
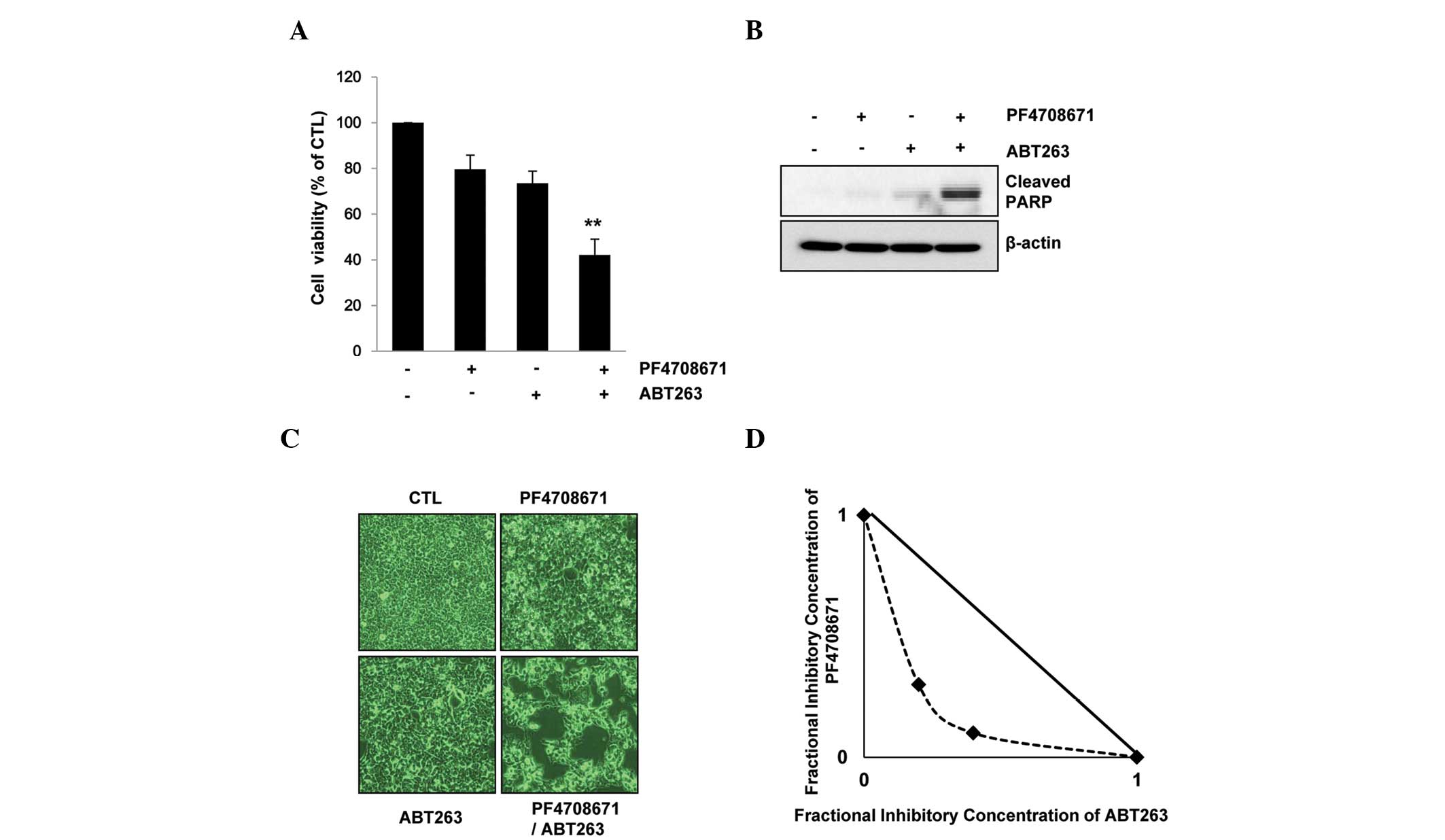
Combined treatment with ABT263 and
PF470867 synergistically induces cell death
The present study examined the effect of combined
treatment with ABT263 and PF4708671 on the induction of cell death.
Cell viability was reduced by 20% following treatment with 10 µM
PF4708671 and by 27% following treatment with 1 µM ABT263. However,
combination treatment with the two agents resulted in a significant
reduction in cell viability (~60%; P<0.01) (Fig. 2A) and the induction of apoptotic cell
death, as examined by PARP cleavage and microscopy images of
apoptosis (Fig. 2B and C).
Furthermore, isobolographic analysis clarified the synergy between
ABT263 and PF4708671 (Fig. 2D). These
data indicate that combined treatment with ABT263 and PF4708671
synergistically induces cell death in BT474 breast cancer
cells.
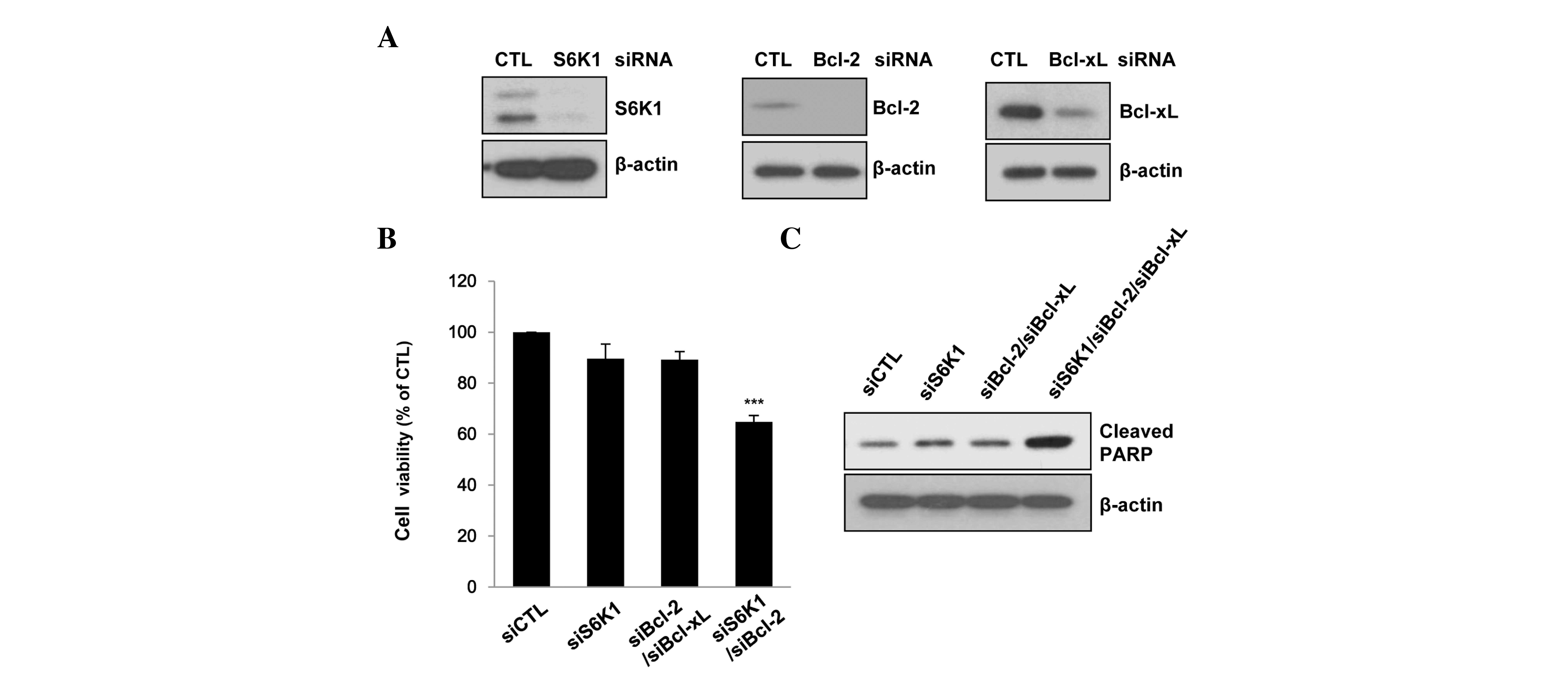
Silencing of S6K1 and Bcl-2/Bcl-xL
induces cell death
To further investigate the effect of the targeting
of S6K1 and Bcl-2/Bcl-xL on BT474 cell death, the inhibitory
effects of siRNAs targeting S6K1 and Bcl-2/Bcl-xL were
investigated. The siRNAs caused marked knockdown of the target
genes (Fig. 3A). The combined siRNA
knockdown of S6K1 and Bcl-2/Bcl-xL significantly inhibited cell
viability and induced PARP cleavage (P<0.001). By contrast, S6K1
or Bcl-2/Bcl-xL siRNA alone did not induce PARP cleavage (Fig. 3B) and exhibited marginal inhibitory
effects on cell viability (Fig. 3C).
These data indicate that the synergistic targeting of S6K1 and
Bcl-2/Bcl-xL may induce BT474 cell death.
Downregulation of survivin expression
by PF4708671 and ABT263 induces cell death
The present study invesitgated the mechanisms
involved in ABT263- and PF4708671-induced cell death. Combined
treatment with ABT263 and PF4708671 appeared to have no effect on
the expression of Bcl-2-related proteins, such as Bcl-2, Bcl-xL and
Mcl-1. Notably, survivin protein, unlike inhibitor of apoptosis
(IAP) family members, such as X-linked IAP (XIAP) and cellular IAP
1 (cIAP1), was significantly decreased in the presence of ABT263
and PF4708671 (Fig. 4A). However,
combined treatment with ABT263 and PF4708671 did not modulate
survivin mRNA expression levels (Fig.
4B), indicating that the observed changes in survivin
expression following ABT263 and PF4708671 exposure did not involve
transcriptional regulation.
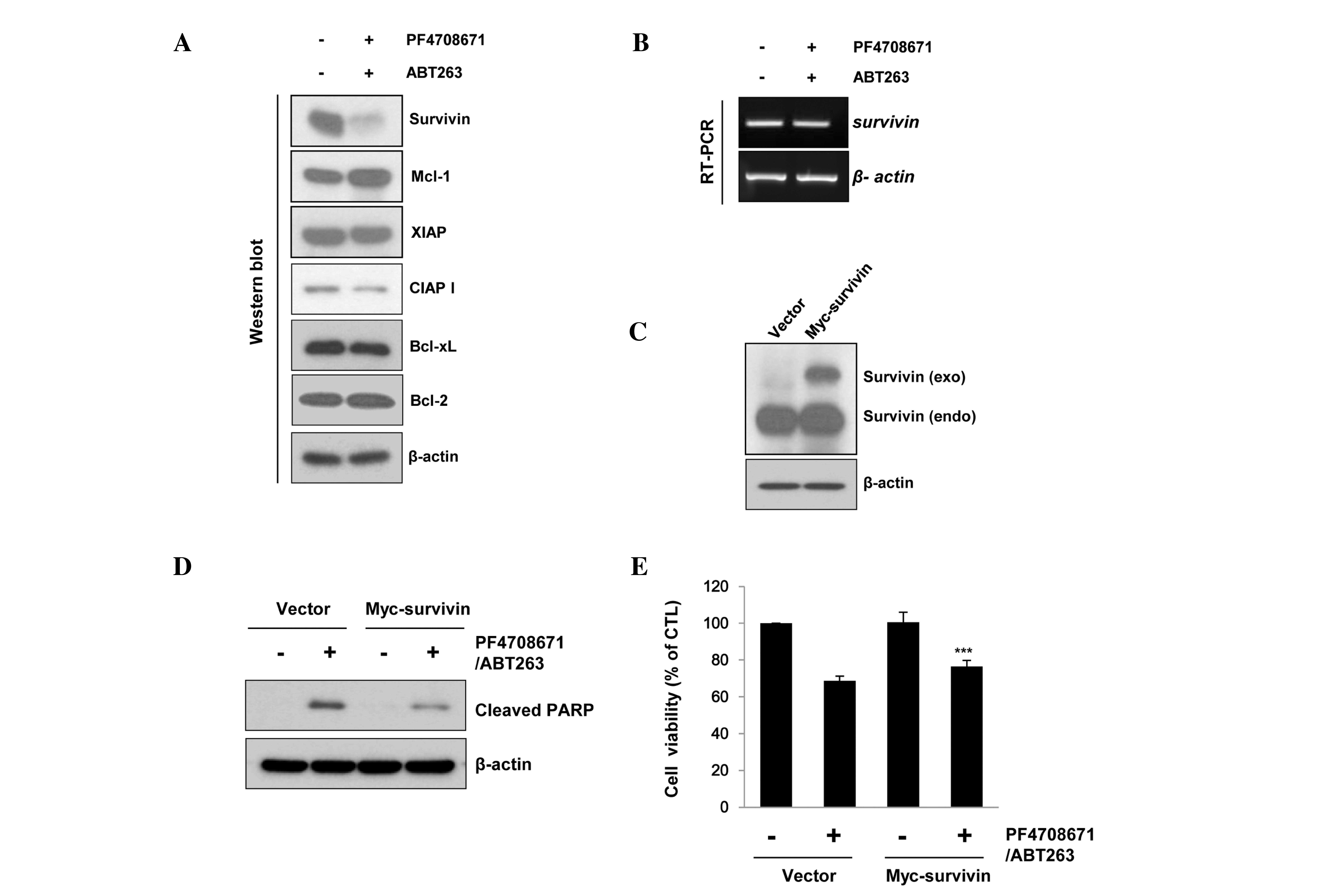 | Figure 4.Downregulation of survivin expression
by PF4708671 and ABT263 induces cell death. (A and B) BT474 cells
were treated with 10 µM PF4708671 and 1 µM ABT263 for 24 h. (C–E)
BT474 cells were transiently transfected with empty or myc-survivin
vector for 24 h, prior to treatment with 10 µM PF4708671 and 1 µM
ABT263 for 24 h. (A, C and E) Protein expression levels were
determined by western blotting and (B) mRNA expression levels were
determined by RT-PCR. (D) Cell viability was analyzed by performing
a 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide
assay. The data are presented as the means of triplicate samples
and the error bars indicate the standard deviation from the mean.
***P<0.001 vs. vector/PF4708671/ABT263-treated group. Mcl-1,
myeloid cell leukemia-1; XIAP, X-linked inhibitor of apoptosis;
cIAP1, cellular inhibitor of apoptosis 1; Bcl-2, B-cell lymphoma-2;
Bcl-xl, B-cell lymphoma-extra large; RT-PCR, reverse
transcription-polymerase chain reaction; CTL, control; PARP,
poly(ADP-ribose) polymerase. |
To further clarify the role of survivin in ABT263
and PF4708671-induced cell death, empty or myc-survivin vectors
were transiently transfected into treated BT474 cells prior to
receiving combined ABT263 and PF4708671 treatment. The expression
of myc-tagged survivin in the BT474 cells significantly suppressed
the cell cytotoxicity induced by PF4708671 and ABT263 (P<0.001;
Fig. 4C and D), and attenuated the
PARP cleavage induced by these inhibitors (Fig. 4E). These data indicate that the cell
death induced by PF4708671 and ABT263 is at least in part mediated
by the downregulation of survivin protein.
Discussion
The impairment of apoptosis is a hallmark of cancer
and is key in the resistance to anticancer therapy. Tumors deploy
diverse strategies to limit or evade apoptosis, frequently
involving perturbation of the intrinsic Bcl-2 apoptotic pathway
(19). As the overexpression of the
pro-survival protein Bcl-2 is common in breast cancer (6), Bcl-2 may serve as a potential target for
the treatment of this disease. ABT263 is an orally bioavailable
small molecule inhibitor of Bcl-2 family proteins that binds with
high affinity to Bcl-2 and Bcl-xL, and antagonizes their
anti-apoptotic function (20). In the
present study, ABT263 (≤2 µM) treatment resulted in upregulated
Mcl-1 expression, in agreement with a previous report (21). However, ABT263 (≤2 µM) did not induce
apoptotic cell death in BT474 breast cancer cells (Fig. 1). Preclinically, ABT263 has
demonstrated limited activity in the majority of solid tumors
containing breast cancer (22–24).
However, when used in combination with other agents, such as
rapamycin, rituximab, rituximab-cyclophosphamide, doxorubicin,
vincristine and prednisolone, and bortezomib, ABT263 appeared to
enhance their efficacy in leukemia and lymphoma models (23).
Our previous study indicated that phosphorylated
S6K1, a downstream effector of mTORC1, may be effectively applied
as a predictive marker for breast cancer cells (25). Indeed, S6K1 is frequently
overexpressed in breast cancer cells (19). In the present study, although
PF4708671 (an S6K1 inhibitor) suppressed S6K1 activity in the BT474
cells, PF4708671 (≤20 µM) did not result in cell death (Fig. 1), indicating that PF4708671 alone is
not sufficient to cause apoptosis. However, it was identified that
combination treatment with ABT263 and PF4708671 synergistically
induced BT474 cell death (Fig. 2).
Furthermore, the combined inhibition of Bcl-2, Bcl-xL and S6K1
using siRNAs induced marked apoptosis (Fig. 3). These data indicate that ABT263 and
PF4708671 in combination may be particularly effective treatment
strategy for breast tumors with high levels of Bcl-2 and S6K1
expression.
ABT263 and PF4708671 combination treatment had no
effect on the expression on anti-apoptotic Bcl-2 family proteins,
such as Bcl-xL and Bcl-2, or IAP family proteins, such as XIAP and
cIAP. However, ABT263 and PF4708671 notably induced decreases in
survivin protein expression without affecting its mRNA expression.
Furthermore, the overexpression of survivin attenuated the cell
death induced by ABT263 and PF4708671. These results indicate that
survivin may be involved in regulating the apoptosis induced by
ABT263 and PF4708671.
In conclusion, data obtained in the present study
indicates that ABT263 or PF4708671 treatment alone cannot induce
apoptosis in breast cancer BT474 cells. However, combined treatment
with ABT263 and PF4708671 appears to extensively induce cell death.
Furthermore, the downregulation of survivin by ABT263 and PF4708671
treatment may induce cell death. Therefore, Bcl-2 and S6K1, which
are frequently activated in breast cancer, may serve as potential
targets in the treatment of patients with breast cancer.
Acknowledgements
The present study was supported by a grant from the
Radiation Bio-Resource Research Program of the Korea Institute of
Radiological and Medical Sciences (grant no. 740802).
References
|
1
|
Strasser A, Harris AW, Bath ML and Cory S:
Novel primitive lymphoid tumours induced in transgenic mice by
cooperation between myc and bcl-2. Nature. 348:331–333. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng J, Carlson N, Takeyama K, Dal Cin P,
Shipp M and Letai A: BH3 profiling identifies three distinct
classes of apoptotic blocks to predict response to ABT-737 and
conventional chemotherapeutic agents. Cancer Cell. 12:171–185.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Callagy GM, Pharoah PD, Pinder SE, et al:
Bcl-2 is a prognostic marker in breast cancer independently of the
Nottingham Prognostic Index. Clin Cancer Res. 12:2468–2475. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dawson SJ, Makretsov N, Blows FM, et al:
BCL2 in breast cancer: A favourable prognostic marker across
molecular subtypes and independent of adjuvant therapy received. Br
J Cancer. 103:668–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tse C, Shoemaker AR, Adickes J, et al:
ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor.
Cancer Res. 68:3421–3428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rudin CM, Hann CL, Garon EB, et al: Phase
II study of single-agent navitoclax (ABT-263) and biomarker
correlates in patients with relapsed small cell lung cancer. Clin
Cancer Res. 18:3163–3169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kamal A, Faazil S and Malik MS:
Apoptosis-inducing agents: A patent review (2010–2013). Expert Opin
Ther Pat. 24:339–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong SE, Kim EK, Jin HO, et al: S6K1
inhibition enhances tamoxifen-induced cell death in MCF-7 cells
through translational inhibition of Mcl-1 and survivin. Cell Biol
Toxicology. 29:273–282. 2013. View Article : Google Scholar
|
|
11
|
Bärlund M, Forozan F, Kononen J, et al:
Detecting activation of ribosomal protein S6 kinase by
complementary DNA and tissue microarray analysis. J Natl Cancer
Inst. 92:1252–1259. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brugge J, Hung MC and Mills GB: A new
mutational AKTivation in the PI3K pathway. Cancer Cell. 12:104–107.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sinclair CS, Rowley M, Naderi A and Couch
FJ: The 17q23 amplicon and breast cancer. Cancer Res Treat.
78:313–322. 2003. View Article : Google Scholar
|
|
14
|
Kim EK, Kim JH, Kim HA, et al:
Phosphorylated S6 kinase-1: A breast cancer marker predicting
resistance to neoadjuvant chemotherapy. Anticancer Res.
33:4073–4079. 2013.PubMed/NCBI
|
|
15
|
Jin HO, Yoon SI, Seo SK, et al:
Synergistic induction of apoptosis by sulindac and arsenic trioxide
in human lung cancer A549 cells via reactive oxygen
species-dependent down-regulation of survivin. Biochem Pharmacol.
72:1228–1236. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu TS, Yiao SY, Lim K, et al:
Interpretation of biological and mechanical variations between the
Lowry versus Bradford method for protein quantification. N Am J Med
Sci. 2:325–328. 2010.PubMed/NCBI
|
|
17
|
Jin HO, Lee YH, Kim HA, et al: Inhibition
of vacuolar H+ ATPase enhances sensitivity to tamoxifen via
up-regulation of CHOP in breast cancer cells. Biochem Biophys Res
Commun. 437:463–468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Noh WC, Mondesire WH, Peng J, et al:
Determinants of rapamycin sensitivity in breast cancer cells. Clin
Cancer Res. 10:1013–1023. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Strasser A, Cory S and Adams JM:
Deciphering the rules of programmed cell death to improve therapy
of cancer and other diseases. EMBO J. 30:3667–3683. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vaillant F, Merino D, Lee L, et al:
Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen
receptor-positive breast cancer. Cancer Cell. 24:120–129. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang B, Ni Z, Dai X, et al: The Bcl-2/xL
inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein
in hepatocellular carcinoma cells. Mol Cancer. 13:982014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shoemaker AR, Mitten MJ, Adickes J, et al:
Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small
cell lung cancer xenograft models. Clin Cancer Res. 14:3268–3277.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ackler S, Xiao Y, Mitten MJ, et al:
ABT-263 and rapamycin act cooperatively to kill lymphoma cells in
vitro and in vivo. Mol Cancer Ther. 7:3265–3274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lock R, Carol H, Houghton PJ, et al:
Initial testing (stage 1) of the BH3 mimetic ABT-263 by the
pediatric preclinical testing program. Pediatr Blood Cancer.
50:1181–1189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim EK, Kim HA, Koh JS, et al:
Phosphorylated S6K1 is a possible marker for endocrine therapy
resistance in hormone receptor-positive breast cancer. Breast
Cancer Res Treat. 126:93–99. 2011. View Article : Google Scholar : PubMed/NCBI
|