When cells face external adverse stress, including
glucose depletion, hypoxia and cytotoxicity agents, they respond to
these stresses by adaptive autophagy and endoplasmic reticulum (ER)
stress to restore homeostasis. Recently, two cell biology
processes, autophagy and ER stress, have received extensive
attention. A mounting body of knowledge has demonstrated that ER
stress and autophagy are central to determining cell fate (1–3). Although
the two processes have both been implicated in various human
diseases (4–6), the crosstalk between autophagy and the
unfolding protein response (UPR) remains poorly understood.
Recently, autophagy and UPR have been implicated in malignant cell
survival and drug resistance (7–9). The
present review focusses on the detailed molecular mechanism of the
interplay between ER stress and autophagy. In addition, the effects
of ER stress-mediated autophagy on tumor survival and drug
resistance are also presented.
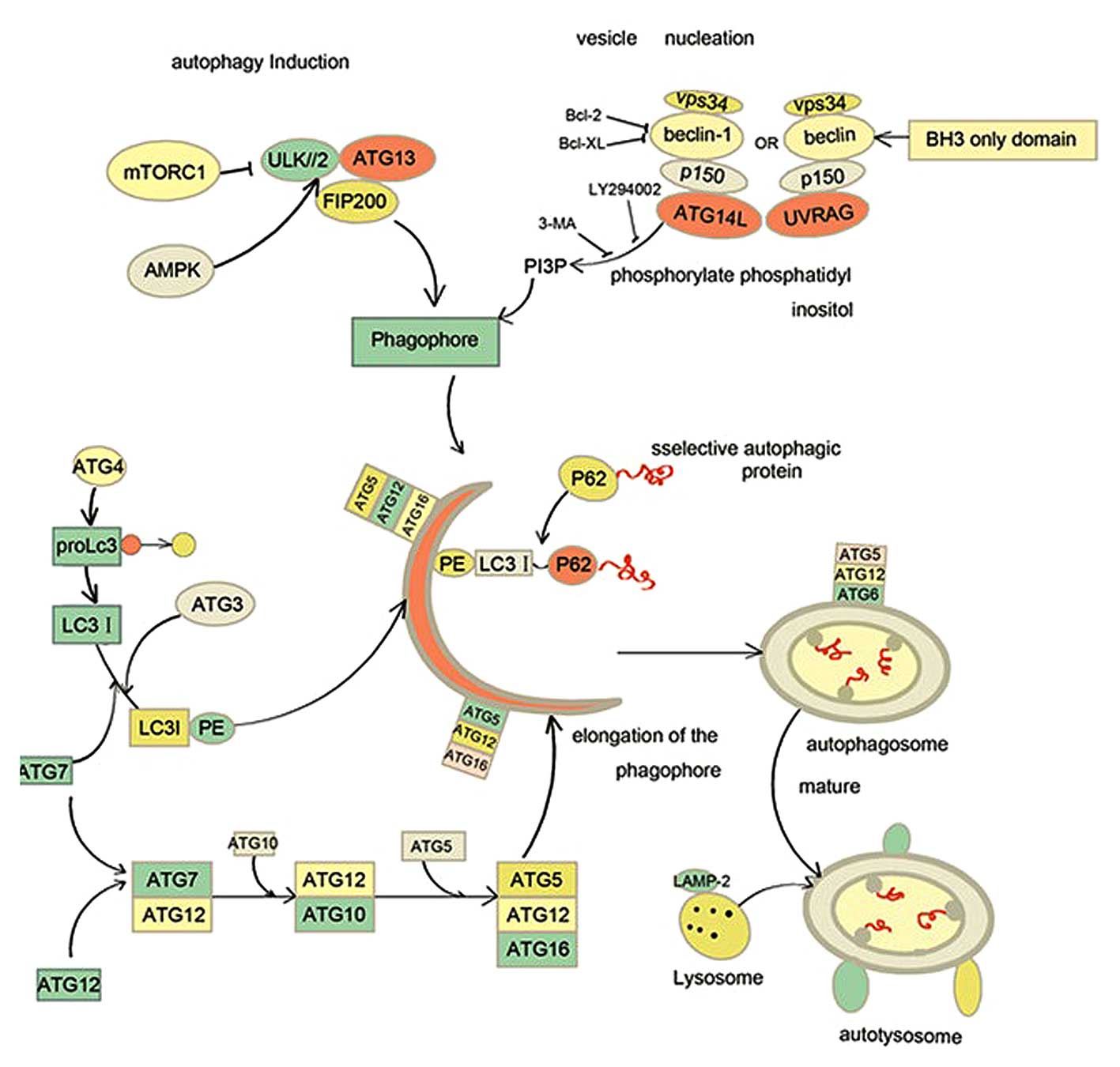
Autophagy is a conserved pathway involving lysosomal
degradation of long-lived proteins or damaged organelles such as
mitochondria and endoplasmic reticulum. Autophagy may promote cell
viability or contribute to cell death, depending on the cell types
and contexts (10). Autophagy is
composed of macroautophagy, chaperone-mediated autophagy and
microautophagy (11,12). Microautophagy involves engulfing
disrupted cellular organelles directly for lysosomal degradation.
In chaperone-mediated autophagy, the cargoes are recognized and
marked by chaperone peptide HSC70 and the substrates are
translocated to lysosomes or endosomes. In macroautophagy,
ubiqiuitinated proteins are identified via adaptors such as p62,
NBR1 (neighbor of BRCA1 gene 1), HDAC6 (Histone Deacetylase 6) and
Afly (11). Macroautophagy is
characterized with formation of double-membraned vesicles to
sequester its cargoes, namely autophagosome. Subsequently, the
autophagosomes fuse with lysosomes or endosomes where the cargoes
are degraded for recycling (13). The
detailed mechanism underlying the process of macroautophagy and its
role in tumor progression and growth have been investigated in
recent years. As the process of macroautophagy has been studied in
the most detail, the present review refers to macroautophagy as
autophagy and predominantly discusses macroautophagy.
In yeast, autophagy is orchestrated by a series of
evolutionarily-conserved autophagy-related genes (ATGs), of which
there are >30 (14). The ATGs make
a significant impact on different stages of autophagy, including
autophagy induction, vesicle nucleation, autophagosomal elongation
and eventual maturation (14).
Mammalian orthologues of the ATGs have also been discovered. The
ULK/ATG13/FIP200 complex is required for the induction of autophagy
in osteosarcoma and NIH3T3 cells (15,16). This
complex is regulated by mammalian target of rapamycin complex 1
(mTORC1). mTORC1 inhibits the ULK/ATG13/FIP200 complex by
phosphorylation of ULK1/2 and ATG13, which suppresses the
phoshorylation of FIR2000 and activity of ULK1/2-ATG13-FIP200
complex. Under starvation stress, adenosine 5′-monophosphate
activated protein kinase (AMPK) senses the low level of glucose or
the low adenosine-triphosphate (ATP)/adenosine monophosphate (AMP)
ratio. Activated AMPK then phophorylates and disrupts the TSC1
(tuberous sclerosis complex 1)/TSC2 (tuberous sclerosis complex 2)
complex, leading to blockage of mTORC1 and autophagy induction
(17). Class III phosphatidylinositol
3-kinase (vps34) combines with beclin-1, p150 and ATG14L to form
the class III PI3PK complex (PI3KC3), which generates
phosphatidylinositol 3-phosphate to allow recruitment of LC3 and
further formation of the autophagosome (18). PI3KC3 has two counterparts, UV
radiation resistance-associated genes (UVRAG) complex and rubicon
complex. The UVRAG complex consists of Vps34, beclin-1, UVRAG and
p150, which contributes to autophagosome maturation (19). By contrast, the rubicon complex is
composed of Vps34, beclin-1, rubicon and p150 and disrupts
autophagosome maturation (20). This
procedure is also suppressed by pharmacological inhibitors of PI3K
complex such as 3-methyladenine (3-MA), LY 294002 and wortmanin. In
addition, anti-apoptotic proteins such as B-cell lymphoma-2 (Bcl-2)
interact with beclin1 and inhibit the nucleation of autophagosome.
c-Jun N-terminal protein kinase 1 (JNK1) and death
associated protein kinase (DAPK) phosphorylate Bcl-2 and positively
regulate autophagy (21,22).
Two ubiquitin-like conjugation systems are essential
for autophagosomal elongation: the ATG12-ATG5 conjugation system
and the yeastATG8/mammalian microtubules associated protein 1 light
chain 3-β (LC3) conjugation system (14,23). In
the former system, Atg12 is covalently conjugated to ATG5 and
physically interacts with ATG16L, which is mediated by E1-like
enzyme ATG7 and E2-like enzyme ATG10 respectively. The
ATG12-ATG5-ATG16L complex acts as a platform to enroll the LC3 II
to the isolated membrane (24). LC3
is cleaved at the C-terminal region to produce LC3 I by ATG4 and
then E1-like enzyme ATG7 and E2-like enzyme ATG3 mediate LC3 I
conjugation with phosphatidylentanolmine to generate LC3 II, the
autophagic membrane form (14).
Subsequently, the selected engulfed cargo is ubiquitinated and
identified by autophagic adaptors, p62/SQSTM1 and neighbor of BRCA1
gene 1 (NBR1). P62 and NBR1 have a similar C-terminal
Ubiquitin-binding domain to bind to ubiquitinated cargo and the LC3
interacting region interacts with LC3 (25,26). The
last machinery of autophagy is the fusion of autophagosome and
lysosomes (Fig. 1). This procedure is
dependent on the binding of LAMP1/2, transmembrane proteins present
in lysosomes. Once an autophagosome fuses with a lyosome, the
cargoes within the autophagosome are degraded for recycling or
digested by acid hydrolases and cathepsins in the lysosomal lumen
(27).
The endoplasmic reticulum (ER) is a critical
cellular organelle for the quality control of secretory proteins:
The ER assists with the correct folding of secreted proteins and
polypeptide chains and transmembrane proteins (28). It also contributes to lipid
biosynthesis and serves as a site for intracellular Ca2+ storage
(29,30). Nascent polypeptide is delivered into
the ER lumen and undergoes the correct posttranslational
modifications and folding in order to perform their functions
efficiently (31). When the protein
quality control procedure is well orchestrated, the correctly
folded secretory proteins or transmembrane proteins are transferred
away from the ER to intracellular apartments or extracellular sites
to execute their roles. However, when misfolded or unfolded
proteins are in abundance, the accumulation of these proteins
triggers ER stress and the UPR ensues (32). The UPR involves an increase in protein
folding capacity as well as reducing the unfolded protein load in
the ER (33). The UPR triggers a
series of cellular processes to recover ER homeostasis: i) The PERK
(protein kinase RNA-like kinase) pathway attenuates the load of
nascent protein in the ER via the global suppression of protein
translation (34); ii) activating
transcription factor 6 (ATF6) and inositol-requiring enzyme 1
(IRE1) together improve the protein folding capacity of the ER by
upregulating the expression of chaperones and foldase enzymes,
which are essential for protein folding (35–37); and
iii) IRE1 signaling facilitates ER associated degradation (ERAD) to
degrade misfolded proteins and regulate IRE1-dependent decay of
mRNA (RIDD) to cleave RNA essential for ER homeostasis (37,38). If
the UPR fails to restore ER homeostasis, prolonged and severe ER
stress may transform the adaptive UPR response, which protects
cells from death in adverse stress, to deadly output by activating
the ER-dependent apoptosis signaling pathway (36,39–41).
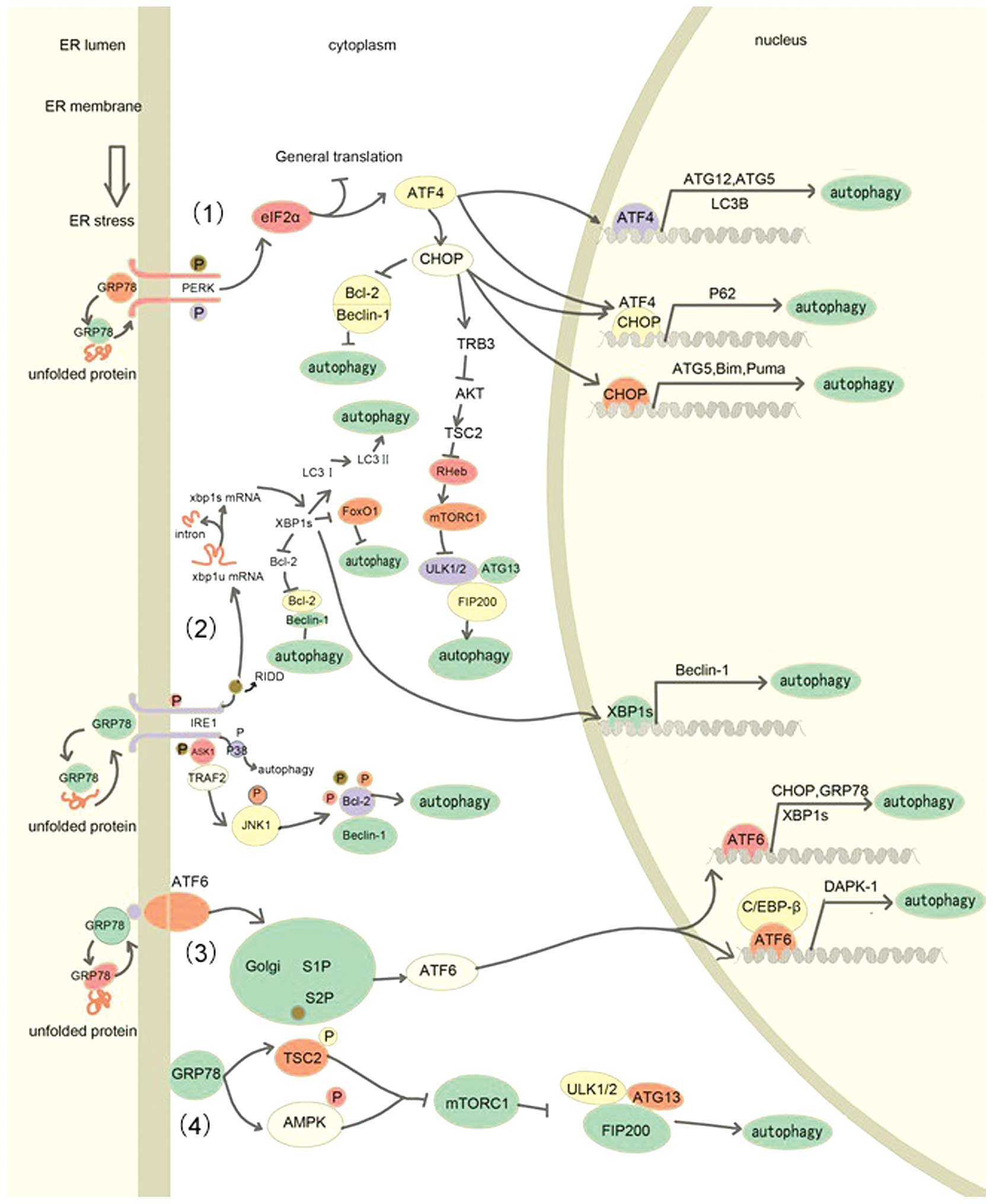
In unstressed conditions, the three transmembrane
proteins (PERK, ATF6 and IRE10) are maintained in an inactive state
by being sequestrated to glucose regulated protein 78 (GRP78), a
light weight chemical chaperone that facilitates protein folding
(Fig. 2). During the UPR, the
increasing concentration of unfolded protein in the ER lumen
completely binds to GRP78 and results in PERK, ATF6 and IRE1
dissociation from GRP78, releasing its inhibitory role on the three
sensors. Subsequently, the three UPR sensors are activated and
initiate adaptive signal transduction through homodimerization and
autophosphorylation (42,43).
That autophagy and ER stress are implicated in cell
fate determination has drawn particular attention (2,8,42). However, the detailed molecular
mechanism for interplay between autophagy and ER stress remains
elusive. In the next section the crosstalk between autophagy and ER
stress is discussed.
JNK belongs to the mitogen-activated protein kinase
(MAPK) super family, which is involved in numerous processes and
has been identified as a ‘stress-associated protein’ (51,52). Upon
initiation of the UPR, IRE1 is activated and recruits the adaptor
protein tumor necrosis factor receptor-associated factor-2 (TRAF2)
to form the IRE1-TRAF2 complex (53).
Subsequently, apoptosis-signal regulating kinase 1 (ASK1) is
enrolled to generate the IRE1-TRAF2-ASK1 complex (54). In the cytosolic facet of ER membrane,
JNK is phosphorylated by the serine/threonine kinase domain of
IRE1. It has been demonstrated that ER stress inducers such as
tunicamycin and thapsigargin can trigger the formation of
autophagic vacuoles and accumulation of LC3-positive vesicles in
mouse embryonic fibroblast cells (MEFs). ER stress-induced
autophagy is dependent on the IRE1/TRAF-2/JNK1 pathway; this is
supported by the observation that MEFs that are deficient in
IRE1/TRAF-2 display markedly reduced formation of autophagosomes
(55). Moreover, SP600125, a
pharmacological inhibitor of JNK1, also blocks the formation of
autophagosomes (52). Yong et
al (56) demonstrated that JNK1
contributed to starvation-induced autophagy via phosphorylating ER
localized Bcl-2 at multi-residues T69, S70 and S87A, leading to
beclin-1 dissociating from ER-localized Bcl-2 and initiating
autophagy (56). In addition, the
JNK1 pathway was demonstrated to serve a pivotal role in regulating
beclin-1 expression at the transcriptional level following
ceramide-induced autophagy in human CNE2 and Hep3B cancer lines
(52). Ceramide-induced upregulation
of beclin-1 and the formation of autophagosomes was inhibited by
SP600125, a specific JNK1 inhibitor and the same phenomenon was
observed using a small interfering RNA targeting JNK mRNA.
Moreover, chromatin immunoprecipitation and luciferase reporter
analysis verified that c-jun, a target of JNK1, was activated and
directly bound to the beclin-1 promoter in ceramide-treated cancer
cells. In this context, the IRE1/JNK1/c-jun pathway is another
important mechanism for autophagy induction. It must be noted that
the IRE1/XBP1s and IRE1/JNK1 induced-autophagy pathways converge at
beclin-1, an ATG protein that is vital during vesicle nucleation.
Targeting beclin-1 may be a novel therapeutic strategy to reverse
the dysfunction of ER stress-induced autophagy in diseases,
including cancer, neurodegenerative disease and diabetes mellitus
(57,58).
Once released from GRP78, PERK, a serine/threonine
kinase, is activated through autophosphorylation and
homodimerization. The activated PERK phosphorylates eIF2α at serine
51, disrupting the assembly of initiator Met-tRNA and the ribosome,
resulting in the suppression of general protein synthesis (59,60).
Paradoxically, phosphorylation of eIF2α promotes the translation of
ATF4, which enhances the ER's capacity for protein folding
(61–63). Subsequently, increased level of ATF4
promotes the translation of CCAAT/enhancer binding protein (C/EBP)
homologous protein (CHOP), which acts as a marker to detect the
induction of the UPR and is involved in the ER stress-mediated
apoptotic pathway (64,65).
It has been demonstrated that polyglutamine (72Q)
aggregates induce vesicular formation and conversion of LC3 is
dependent on PERK/eIF2α activation. This is supported by the fact
that 72Q-mediated induction of autophagy is suppressed
significantly in MEF cells with an eIF2αA/A mutation or in MEF
cells transfected with PERK (dominant-negative PERK) (66). MEF cells with an eIF2αA/A mutation
cannot be phosphorylated by PERK, and dominant-negative PERK MEF
cells cannot phosphorylate eIF2αA and can prevent wild-type PERK
phosphorylating eIF2αA. In addition, ATF4 is responsible for
upregulation of ATG12 (67), a key
component of the Atg5-Atg12-Atg16L complex, which is essential for
the elongation of autophagosomes. Similarly, BRAF inhibition
induces phosphorylation of PERK and is crucial for autophagosome
formation in melanoma (68). Blockage
of PERK using either the pharmacological inhibitor GSK2606414, or
an siRNA against PERK elicits a marked reduction in the LC3 II/I
ratio. Furthermore, ATF4 directly binds to the cyclic AMP response
element binding site in the promoter of microtubule-associated
protein 1 light chain 3 β (LC3β), a vital constituent of
autophagosomal membrane, and promotes expression of LC3β, which
facilitates the induction of autophagy (67,69).
CHOP is a potent transcription factor, which is
implicated in autophagy induction. This transcription factor has
been implicated in various cellular processes, including
proliferation, differentiation, apoptosis, autophagy and the UPR
(70–72). It has been demonstrated that CHOP
expression increases the expression levels of ATG5 and BH3-only
protein (73,74), in addition to reducing the expression
levels of Bcl-2, which contributes to beclin-1 releasing from Bcl-2
(72,75). In addition, CHOP expression also
results in increased expression levels of BH3-only proteins such as
Bim and Puma, which also bind to Bcl-2 via the single BH3 domain
and this further releases beclin-1 from the Bcl-2-beclin-1 complex
(74). Furthermore, the PERK-CHOP
pathway provokes the induction of tribbles-related protein3 (TRB3),
which blocks the activation of protein kinase B (Akt) (76). TRB3-mediated inhibition of Akt
attenuates the phosphorylation of TSC2 (tuberous sclerosis complex
2) on serine/theronine residues, leading to an inhibitory regulator
of Ras homolog enriched in brain (Rheb) and inactivation of mTORC1.
Finally, inactivation of mTORC1 dephosphorylates ATG13 and the
ULK1/2 complex and initiates autophagosomal formation (77). The eIF2α/ATF4/CHOP axis promotes the
expression of p62 at the transcriptional level through binding to
the AARE sequence of p62 promoter to regulate autophagy induction
(78).
ATF6 acts as an ER transmembrane sensor,
characterized by its C terminus in the ER lumen and N-terminus
(possessing the transcription factor activity) in the cytosol
(79). When the amount of misfolded
protein increases to the threshold, ATF6 escapes from the
sequestration of GRP78 and exposes the Golgi localization signals
to facilitate its delivery to the Golgi apparatus. ATF6 undergoes
cleavage by Golgi apparatus localized site 1 and site 2 proteases
(80). Subsequently, the activated
ATF6 is translocated to nucleus and then binds to ER
stress-associated elements. ATF6 elevates the expression of GRP78,
GRP94, XBP1, CHOP and protein disulfide isomerase, which is
essential for assisting correct protein folding and secretion
(81–83). It has previously been demonstrated
that ATF6 is required for induction of autophagy. Death-associated
kinase 1 (DAPK1) is involved in ATF6 mediated autophagy (84). The mechanism underlying ATF6 induced
autophagy is that ATF6 interacts with C/EBP-β to form a
transcriptional heterodimer complex and then binds to CRE/ATF
elements of the DAPK1 promoter to induce the expression of
DAPK1. Knockdown of ATF6 with specific shRNAs and cells with
ATF6−/− displayed strongly reduced expression of DAPK1
and reduced autophagosome formation. Indeed, DAPK1 has been
implicated in driving autophagosome formation through
phosphorylation of beclin-1 (85).
Meanwhile, ATF6-mediated upregulation of CHOP, XBP1 and GRP78 also
contributes to ATF6-induced autophagy (1). This indirect pathway adds a further
layer of complexity in ER stress-induced autophagy.
GRP78 is a key UPR trigger and ER molecular
chaperone, which has been demonstrated to induce autophagy.
Knockdown of GRP78 suppresses autophagy. However, the
siGRP78-dependent autophagy inhibition was reversed following the
addition of siXBP-1 (86).
Accumulating evidence has demonstrated that autophagy relies on
intact ER function and its correct morphology, which provides an
essential membrane for autophagosomal elongation and nucleation.
Knockdown of GRP78 disrupting normal ER function and morphology may
be attributed to the suppression of ER stress-induced autophagy
(86). This finding was also strongly
supported by the observation that GRP78 induced activation of AMPK
and TSC2, which results in the inhibition of mTOR and induction of
autophagy in breast cancer (87).
In fibroblasts from patients with Pompe disease,
accumulation of misfolded acid α-glucosidase (GAA) induced ER
stress and resulted in increased levels of LC3 II and autophagosome
formation. Mechanistically, the activation of p38 MAPK signaling
pathways were essential for this phenomenon (88). NB-DNJ, a pharmacological chaperone for
misfolded GAA, dramatically reduces the level of p38
phosphorylation and p38-associated ER stress. The autophagic flux
induced by ER stress was also attenuated following treatment with
SB203580, a specific p38 MAPK inhibitor. Similarly, another study
uncovered an increase in p38 phosphorylation and induction of
autophagy in human gingival cells after exposure to ER stress
agents brefeldin A, thapsigargin and tunicamycin (89). In this context, SB203580 suppressed ER
stress-induced autophagy. As a downstream target of ER stress, p38
was demonstrated to be phosphorylated by the IRE1/ASK1 axis.
Notably, JNK, another MAPK, is known to be a common target of the
IRE1/ASK1/TRAF2 pathway. However, the level of phosphorylated JNK
and ERK remained unchanged in fibroblasts and human gingival cells
in response to ER stress. Therefore, which pathway among the three
MAPK pathways is the preferential mediator to induce autophagy in
ER stress condition appears elusive at present.
Autophagy alleviated ER stress may also be
established, which completes the feedback loop of crosstalk between
autophagy and ER stress (90).
Normally, ER stress induces a process that delivers misfolded
proteins to the cytoplasm where they are ubiquitinated and degraded
by the ubiquitin-proteasome system, which is termed ER associated
degradation (ERAD) (91). However,
when the process of ERAD is saturated or disrupted, ER
stress-induced autophagy is considered to degrade the insoluble
misfolded or unfolded proteins to alleviate the ER stress and
recover homeostasis (92,93). This hypothesis is supported by a study
that reported that HCT116 and DU145 cells displayed increased
levels of ER stress following impairment of autophagy via a
pharmacological inhibitor or the transfection of an siRNA targeting
BECLIN-1 or LC3B (92). Furthermore,
autophagy may counterbalance ER expansion by sequestering the ER
into double membrane-bounded and autophagosomal-like structures
(94). Rapamycin, a well-established
autophagy inducer, has been observed to reduce
hypoxia/ischemia-induced ER stress significantly in vivo
(90). In addition, 3-methyladenine,
an early pharmacological inhibitor of autophagy, completely
reverses the inhibition of ER stress (90).
Previous studies have indicated that autophagy and
ER stress protect cancer cells exposed to various stresses from
death (57,95). Osteosarcoma cells display increased
levels of autophagosomal formation when treated with anticancer
agents, including cisplatin, doxorubicin and methotrexate (96). The autophagy induced by anticancer
agents is suppressed by 3-MA or knockdown of beclin-1, ATG7 or
PI3KC3, which may sensitize the osteosarcoma to the anticancer
agents. These finding indicate that autophagy in response to
anticancer agents may contribute the chemotherapeutic resistance of
osteosarcoma (96,97). The latest findings about autophagy and
ER stress mediated pro-survival and drug resistance in malignancies
are now presented.
The molecular mechanism for GRP78 protecting tumor
cells against chemotherapeutic agents as established from previous
studies is as follows (Fig. 3): i)
GRP78 binds and inactivates the pro-apoptotic protein caspase-7,
which is localized in the ER outer membrane (103,104).
GRP78 simultaneously mediates CHOP suppression and reduces
CHOP-dependent apoptosis. ii) It has been proposed that GRP78 may
bind to BIK through its BH-3 domain, which reverses the increased
BIK expression in breast cancer that results from the presence of
anti-estrogen agents, disrupting BIK/BCL-2 complex formation. An
increase of free BCL-2 in the ER membrane then alleviates
Ca2+ leakage, in addition to the release of cytochrome c
from mitochondria to the cytoplasm, blocking caspase-dependent
apoptosis (104,105). iii) Silencing GRP78 inhibits renal
cell carcinoma (RCC) growth and induces G1 cell-cycle arrest.
Knockdown of GRP78-induced activation of the UPR results in a
marked suppression of G1/S translation-associated cyclins (D1, D2
and E2) and cylin-dependent kinase (CKD4 and CKD6) expression
(101). iv) A study has demonstrated
that GRP78 may be delivered to and anchored at the cell surface as
a receptor during ER stress (106).
The cell surface GRP78 activates the PI3K/AKT pathway and interacts
with Cripto to suppress the transforming growth factor-β (TGF-β)
pathway, promoting cell survival and growth (10). This data is supported by a number of
previous studies that have demonstrated that upregulation of GRP78
results in the chemoresistance phenotype of breast cancer,
malignant gliomas and tumor associated endothelia cells (100,107,108).
The characteristics of cancer cells include high
rates of proliferation, insufficient supply of oxygen and glucose,
apoptotic resistance and angiogenesis; therefore, cancer cells
require increased rates of aerobic glycolysis and glutamine
consumption for growth (109).
c-Myc-dependent glutamine metabolism has been implicated in
assisting cancer cell survival during glucose deprivation (110). It has been indicated that elevated
levels of GRP78 induce c-Myc expression and promote
c-Myc-mediated glutamine metabolism, which contributes to
cell survival (111). The mechanism
for enhanced c-Myc-mediated glutamine metabolism is attributed to
GRP78 disrupting adenomatous polyposis coli (APC)-β-catenin and
E-cadherin β-catenin complexes, which results in the extracellular
release of APC and an enhanced level of free β-catenin. Eventually,
the high expression levels of intermediaries of the β-catenin
pathway facilitates the c-Myc-mediated glutamine metabolism. This
finding adds support to the hypothesis that GPR78 acts as a novel
link between metabolic changes and tumor survival. Taken together,
these observation indicate that overexpression of GRP78 occurs in
tumors and confers drug resistance (112). Targeting GRP78 may be a novel
strategy to overcome the barrier of chemotherapeutic failure in the
future.
Under most circumstances, the UPR is considered to
be a cytoprotective response, whose main goal is to reduce the
protein load that requires folding and to increase the capacity for
folding protein in the ER lumen (40). In the three major branches of UPR, the
IRE1 is considered to elicit the pro-survival output. IRE1 has been
demonstrated to be associated with cancer proliferation and
angiogenesis in vitro and in vivo (113). Glioma cells that express
dominant-negative IRE1α display a markedly reduced growth rate and
reduced angiogenic signalling (114). In addition, persistent activation of
IRE1 was also responsible for the resistance of melanoma cells to
ER stress-induced apoptosis (115).
XBP1s and RIDD are two potent IRE1-induced
pro-survival signals that occur in adverse stress (36). IRE1 mediated unconventional splicing
of XBP-1 mRNA and regulation of cyclin A1 expression favors
IRE1-induced cancer cell growth (Fig.
3) (116). Elevated levels of
XBP1 splicing have been observed in various types of tumor and
predict poor outcomes for patients (117,118).
Triple-negative breast cancer (TNBC) is an aggressive tumor with
few effective treatment options characterized by the absence of
estrogen receptor, progesterone receptor and HER2 (human epidermal
growth factor receptor-2) expression but high levels of XBP1s
expression. Impairment of XBP1 splicing markedly inhibits TNBC
growth, metastasis and angiogenesis (119). In addition, xenograft mice
transfected with MDA-MB-231 breast cancer cells with an shRNA
targeting XBP1, reduces the risk of breast cancer tumor relapse
(119). Furthermore, it has been
established that XBP1s binds to hypoxia inducible factor-1α (HIF1α)
via its amino-terminus-bZIP domain and promotes the expression of
HIFα targeting genes, including vascular endothelial growth
factor-A, phosphoinositide-dependent kinase 1, GLUT1 and
DNA-damage-inducible transcript 4, which confer pro-survival
signaling responses to hypoxic stress (119). Similarly, another study indicated
that XBP1s is critical in myeloma pathogenesis and a high ratio of
XBP1s/XBP1 unspliced is closely correlated with poor outcome and a
shortened relapse interval in patients (120). It has also been identified that the
blockage of IRE1α endoribonuclease activity with novel small
molecules such as MKC-3946 and STF-083010, inhibits the splicing of
XBP1 in multiple myeloma (MM) when in untreated condition. In
addition, MKC-3946 treatment also leads to a significant
suppression of XBP1 splicing and enhancement of ER mediated
apoptosis in MM cells when concurrently treated with bortezomib or
17-allylamino-17-demethoxygeldanamycin (17-AGG). Treatment with
either of the two specific IRE1 endoribonuclease inhibitors exerts
no effect on the kinase activity and autophoshorylation of IRE1 but
only marked inhibition of XBP1 splicing and its downstream
substrates, which strongly demonstrates that the IRE1-XBP1 axis is
essential for MM cell survival and targeting this pathway may
result in marked anti-tumor effects (120).
Dimerization and autophophorylation of PERK ensues
following dissociation from GRP78; PERK then phosphorylates eIF2α
at serine 51 and nuclear factor erythroid-2-related factor 2 (NRF2)
(121). Activation of NRF2 promotes
resistance to hypoxia in cells through enhancing the expression of
enzymes that scavenge reactive oxygen species (ROS) (122). Knockdown of PERK sensitizes
esophageal and breast tumor cells to chemotherapeutic agents and
impairs the growth of these two malignant types of cancer in
vitro, which is attributed to the activation of the double
stranded DNA breakage checkpoint to trigger G2/M arrest and the
accumulation of ROS (Fig. 3)
(123). A highly selective PERK
inhibitor, GSK2656157, has been demonstrated to block the ER
stress-induced PERK autophosphorylation and attenuate the
phosphorylation of eIF2α and expression of downstream messengers,
ATF4 and CHOP (124). Furthermore,
treatment with GSK2656157 robustly reduced angiogenesis and altered
amino acid metabolism, which impaired human xenograft tumor growth
in mice (124). Similarly,
ATF4−/− cells demonstrate increased sensitivity to
hypoxic stress (125). Moreover,
another novel mechanism underlying PERK-dependent pro-survival
signaling has been reported by Hamanaka et al. PERK induced
upregulation of cellular inhibitor of apoptosis (cIPA1 and cIPA2)
contributes to the protection of cells against tunicamycin-induced
death (126). Finally, the
PERK-eIF2α axis is robustly elevated in chronic myeloid leukemia
(CML) cells that also express high levels of BCR-ABL (127). Meanwhile, genetic modification of
CML cells via transfection with dominant-negative mutants of PERK
or dominant-negative eIF2α-S51A mutant, results in markedly
increased levels of apoptosis when treated with imatinib (127). Indeed, compromised PERK-eIF2α
phosphorylation significantly extends the population doubling time
and results in smaller clone sizes in human K562 CML cells with
dominant-negative PERK or eIF2α. Collectively, the PERK arm
substantially contributes to the growth of tumor cells and elicits
a dominant pro-survival output in tumor cells when treated with
anti-tumor agents (127). Thus,
targeting the PERK-eIF2α pathway represents another promising
strategy to override the barriers for dealing with malignant
tumors.
The role of ATF6 in tumor chemoresistance has not
been extensively studied at present. However, accumulating evidence
on ATF6-dependent tumor drug resistance has uncovered that ATF6 is
another contributor to cancer drug resistance. The detailed
mechanism for ATF6 activation and ATF6 induced imatinib resistance
in leukemia has been described (128). In this model, protein disulfide
isomerase 5 (PDIA5) was identified as being essential for ATF6
activation and export of proteins from the ER lumen. Ablation of
PDIA5 reduced the expression of ATF6 specific target genes.
Furthermore, silencing of ATF6 expression sensitized K562R cells (a
leukemia cell line resistance to imatinib) to the treatment of
imatinib. In addition, persistent activation of ATF6 and reduced
apoptosis were revealed in tunicamycin or thapsigargin-treated
melanoma. It may therefore be concluded that ATF6 activation is
essential for protecting melanoma against ER stress-induced cell
death (115). In addition to the
roles of ATF6 in cell survival and drug resistance in proliferating
malignant tumor cells, ATF6 mediated pro-survival and
chemoresistance in dormant tumor cells: ATF6 was demonstrated to be
responsible for tumor relapse in the human dormant squamous
carcinoma cell line, D-HEp3 (129).
P38 signaling dependent activation and nuclear localization of
ATF6α has been demonstrated in D-HEp3 by immunoblotting and
immunofluorescence analysis. Moreover, when D-HEp3 cells in which
ATF6α expression has been knocked down are treated with
doxorubicin, the number of viable cells is markedly reduced
(129). The mechanism by which ATF6α
elicits its anti-chemotherapeutic effects in D-HEp3 cells is
considered to be dependent on the activation of mTOR, supported by
the evidence that knockdown of Rheb sensitizes the D-HEp3 to
tunicamycin (129).
Depending on the cell types and context, autophagy
has been considered to promote tumor survival or facilitate tumor
suppression (8,9,130).
Mounting evidence has demonstrated that the UPR induces autophagy
in various types of malignant tumor (131,132). A
number of studies have also strongly indicated that UPR induced
autophagy is critical for malignant tumors cells to survive in
adverse stress (55,133).
Melanoma exhibits elevated levels of ER stress and
autophagy following treatment with the specific BRAF inhibitor,
PLX4720. The BRAF inhibitor induced autophagy relies on
PERK-dependent ER stress. Blockage of the UPR induced autophagy
limits melanoma resistance to the BRAF inhibitor (134). Notably, UPR induced autophagy
promotes survival in HCT116 and DU145 cell lines when exposed to ER
stress inducers (92). By contrast,
suppression of UPR-induced autophagy reduces cell death in primary
colon cells and MEF cells when treated with ER stress inducers
(92). This observation indicates
that UPR induced autophagy exerts a pro-survival response in
malignant tumor cells but elicits a cell death response in normal
cells. Moreover, another study also strongly supports this
hypothesis. The human P493-6B cell line, which expresses high
levels of the oncogene c-Myc, exhibits elevated levels of UPR and
autophagy (135). This c-Myc induced
UPR protects P493-6B cells against c-Myc induced cell death. To
further uncover the mechanism of UPR pro-survival consequence, Hart
et al demonstrated that there is enhanced autophagosome
formation and increased LC3 I/II conversion in P493-6B cells with
high expression levels of c-Myc (135). Meanwhile, PERK ablation attenuates
c-Myc induced autophagy, indicating the critical role of
PERK in autophagy induction. Moreover, disrupting autophagy with
bafilomycin A1 in P493-6B cells increases cell death in response to
c-Myc activation. Therefore, UPR induced autophagy has a
pro-survival role in this context.
The present review describes the core pathway of
autophagy and UPR, in addition to their regulations. Moreover, the
detailed molecular mechanisms underlying the crosstalk between
autophagy and ER stress are discussed. Accumulating evidence
identifies that the three arms of UPR exert marked influences on
the induction of autophagy. In turn, autophagy also counterbalances
ER stress via degradation of protein aggregates and attenuation of
ER expansion. This negative feedback loop allows an insight into
the intrinsic orchestrated pathways in cells under adverse
conditions. In the present review, the roles of UPR induced
autophagy in malignant tumor survival and drug resistance were also
discussed. Targeting the UPR induced autophagy response may guide
novel therapeutic approaches. Given the vital role of the UPR and
autophagy in determining tumor cells fate, further studies on how
to manipulate these cell processes are essential to broaden our
concepts on tumor therapeutic strategies.
The present review was supported by the National
Natural Science Foundation of China (grant no. 81272947).
|
1
|
Mei Y, Thompson MD, Cohen RA and Tong X:
Endoplasmic reticulum stress and related pathological processes. J
Pharmacol Biomed Anal. 1:10001072013.PubMed/NCBI
|
|
2
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan F, Li J, Chen J, Hu Q, Gu C, Lin W and
Chen G: Endoplasmic reticulum stress is associated with
neuroprotection against apoptosis via autophagy activation in a rat
model of subarachnoid hemorrhage. Neurosci Lett. 563:160–165. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimodaira Y, Takahashi S, Kinouchi Y,
Endo K, Shiga H, Kakuta Y, Kuroha M and Shimosegawa T: Modulation
of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP
homologous protein (CHOP) and inositol-requiring enzyme 1α (IRE1α)
in human colon cancer cells. Biochem Biophys Res Commun.
445:524–533. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rubiolo JA, López-Alonso H, Martínez P,
Millán A, Cagide E, Vieytes MR, Vega FV and Botana LM: Yessotoxin
induces ER-stress followed by autophagic cell death in glioma cells
mediated by mTOR and BNIP3. Cell Signal. 26:419–432. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vandewynckel YP, Laukens D, Geerts A,
Bogaerts E, Paridaens A, Verhelst X, Janssens S, Heindryckx F and
Van Vlierberghe H: The paradox of the unfolded protein response in
cancer. Anticancer Res. 33:4683–4694. 2013.PubMed/NCBI
|
|
8
|
Lorin S, Hamaï A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nguyen HG, Yang JC, Kung H, Shi X, Tilki
D, Lara PN Jr, De Vere White RW, Gao AC and Evans CP: Targeting
autophagy overcomes Enzalutamide resistance in castration-resistant
prostate cancer cells and improves therapeutic response in a
xenograft model. Oncogene. 33:4521–4530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klionsky DJ: Autophagy revisited: A
conversation with Christian de Duve. Autophagy. 4:740–743. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
MartinezBorra J and López-Larrea C:
Autophagy and self-defense. Adv Exp Med Biol. 738:169–184. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizushima N: The role of the Atg1/ULK1
complex in autophagy regulation. Curr Opin Cell Biol. 22:132–139.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fader CM, Aguilera MO and Colombo MI:
Autophagy response: Manipulating the mTOR-controlled machinery by
amino acids and pathogens. Amino Acids. Sep 19–2014.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Simonsen A and Tooze SA: Coordination of
membrane events during autophagy by multiple class III PI3-kinase
complexes. J Cell Biol. 186:773–782. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song Z, An L, Ye Y, Wu J, Zou Y, He L and
Zhu H: Essential role for UVRAG in autophagy and maintenance of
cardiac function. Cardiovasc Res. 101:48–56. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun Q, Zhang J, Fan W, Wong KN, Ding X,
Chen S and Zhong Q: The RUN domain of rubicon is important for
hVps34 binding, lipid kinase inhibition and autophagy suppression.
J Biol Chem. 286:185–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maiuri MC, Le Toumelin G, Criollo A, Rain
JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K,
Tavernarakis N, et al: Functional and physical interaction between
Bcl-X (L) and a BH3-like domain in Beclin-1. EMBO J. 26:2527–2539.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakatogawa H, Ichimura Y and Ohsumi Y:
Atg8, a ubiquitin-like protein required for autophagosome
formation, mediates membrane tethering and hemifusion. Cell.
130:165–178. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishida Y, Arakawa S, Fujitani K,
Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y
and Shimizu S: Discovery of Atg5/Atg7-independent alternative
macroautophagy. Nature. 461:654–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsumoto G, Wada K, Okuno M, Kurosawa M
and Nukina N: Serine 403 phosphorylation of p62/SQSTM1 regulates
selective autophagic clearance of ubiquitinated proteins. Mol Cell.
44:279–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rogov V, Dötsch V, Johansen T and Kirkin
V: Interactions between autophagy receptors and ubiquitin-like
proteins form the molecular basis for selective autophagy. Mol
Cell. 53:167–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eskelinen EL: Maturation of autophagic
vacuoles in Mammalian cells. Autophagy. 1:1–10. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Braakman I and Bulleid NJ: Protein folding
and modification in the mammalian endoplasmic reticulum. Annu Rev
Biochem. 80:71–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chistiakov DA, Sobenin IA, Orekhov AN and
Bobryshev YV: Role of endoplasmic reticulum stress in
atherosclerosis and diabetic macrovascular complications. Biomed
Res Int. 2014:6101402014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Beard NA, Laver DR and Dulhunty AF:
Calsequestrin and the calcium release channel of skeletal and
cardiac muscle. Prog Biophys Mol Biol. 85:33–69. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aebi M, Bernasconi R, Clerc S and Molinari
M: N-glycan structures: Recognition and processing in the ER.
Trends Biochem Sci. 35:74–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hiramatsu N, Joseph VT and Lin JH:
Monitoring and manipulating mammalian unfolded protein response.
Methods Enzymol. 491:183–198. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
34
|
Schroder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Iwawaki T, Hosoda A, Okuda T, Kamigori Y,
NomuraFuruwatari C, Kimata Y, Tsuru A and Kohno K: Translational
control by the ER transmembrane kinase/ribonuclease IRE1 under ER
stress. Nat Cell Biol. 3:158–164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Y and Brandizzi F: IRE1: ER stress
sensor and cell fate executor. Trends Cell Biol. 23:547–555. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li M, Baumeister P, Roy B, Phan T, Foti D,
Luo S and Lee AS: ATF6 as a transcription activator of the
endoplasmic reticulum stress element: Thapsigargin stress-induced
changes and synergistic interactions with NF-Y and YY1. Mol Cell
Biol. 20:5096–5106. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maurel M, Chevet E, Tavernier J and Gerlo
S: Getting RIDD of RNA: IRE1 in cell fate regulation. Trends
Biochem Sci. 39:245–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chow SE, Kao CH, Liu YT, Cheng ML, Yang
YW, Huang YK, Hsu CC and Wang JS: Resveratrol induced ER expansion
and ER caspase-mediated apoptosis in human nasopharyngeal carcinoma
cells. Apoptosis. 19:527–541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012.PubMed/NCBI
|
|
41
|
Kim R, Emi M, Tanabe K and Murakami S:
Role of the unfolded protein response in cell death. Apoptosis.
11:5–13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jäger R, Bertrand MJ, Gorman AM,
Vandenabeele P and Samali A: The unfolded protein response at the
crossroads of cellular life and death during endoplasmic reticulum
stress. Biology of the Cell. 104:259–270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hetz C, Martinon F, Rodriguez D and
Glimcher LH: The unfolded protein response: Integrating stress
signals through the stress sensor IRE1α. Physiol Rev. 91:1219–1243.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shajahan AN, Riggins RB and Clarke R: The
role of X-box binding protein-1 in tumorigenicity. Drug News
Perspect. 22:241–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Calfon M, Zeng H, Urano F, Till JH,
Hubbard SR, Harding HP, Clark SG and Ron D: IRE1 couples
endoplasmic reticulum load to secretory capacity by processing the
XBP-1 mRNA. Nature. 415:92–96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gomez BP, Riggins RB, Shajahan AN, Klimach
U, Wang A, Crawford AC, Zhu Y, Zwart A, Wang M and Clarke R: Human
X-box binding protein-1 confers both estrogen independence and
antiestrogen resistance in breast cancer cell lines. FASEB J.
21:4013–4027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Margariti A, Li H, Chen T, Martin D,
VizcayBarrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang
Z, et al: XBP1 mRNA splicing triggers an autophagic response in
endothelial cells through BECLIN-1 transcriptional activation. J
Biol Chem. 288:859–872. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Suzuki H, Kanekura K, Levine TP, Kohno K,
Olkkonen VM, Aiso S and Matsuoka M: ALS-linked P56S-VAPB, an
aggregated loss-of-function mutant of VAPB, predisposes motor
neurons to ER stress-related death by inducing aggregation of
co-expressed wild-type VAPB. J Neurochem. 108:973–985. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vidal RL, Figueroa A, Court FA, Thielen P,
Molina C, Wirth C, Caballero B, Kiffin R, SeguraAguilar J, Cuervo
AM, et al: Targeting the UPR transcription factor XBP1 protects
against Huntington's disease through the regulation of FoxO1 and
autophagy. Hum Mol Genet. 21:2245–2262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li D, Wang L, Deng R, Tang J, Shen Y, Guo
J, Wang Y, Xia LP, Feng GK, Liu QQ, et al: The pivotal role of
c-Jun NH2-terminal kinase-mediated Beclin 1 expression during
anticancer agents-induced autophagy in cancer cells. Oncogene.
28:886–898. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nishitoh H, Matsuzawa A, Tobiume K,
Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H: ASK1
is essential for endoplasmic reticulum stress-induced neuronal cell
death triggered by expanded polyglutamine repeats. Genes Dev.
16:1345–1355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ogata M, Hino SI, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jiang LC, Xin ZY, Deborah B, Zhang JS,
Yuan DY, Xu K, Liu XB, Jiang HQ, Fan QC, Zhang B and Li KY:
Inhibition of autophagy augments apoptosis in human oral squamous
cell carcinoma under nutrient depletion. J Oral Pathol Med.
44:361–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang MZ, Wang Y, Paueksakon P and Harris
RC: Epidermal growth factor receptor inhibition slows progression
of diabetic nephropathy in association with a decrease in
endoplasmic reticulum stress and an increase in autophagy.
Diabetes. 63:2063–2072. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shi Y, Vattem KM, Sood R, An J, Liang J,
Stramm L and Wek RC: Identification and characterization of
pancreatic eukaryotic initiation factor 2 alpha-subunit kinase,
PEK, involved in translational control. Mol Cell Biol.
18:7499–7509. 1998.PubMed/NCBI
|
|
61
|
Lu PD, Jousse C, Marciniak SJ, Zhang Y,
Novoa I, Scheuner D, Kaufman RJ, Ron D and Harding HP:
Cytoprotection by pre-emptive conditional phosphorylation of
translation initiation factor 2. EMBO J. 23:169–179. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Teske BF, Baird TD and Wek RC: Methods for
analyzing eIF2 kinases and translational control in the unfolded
protein response. Methods Enzymol. 490:333–356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wek RC and Cavener DR: Translational
control and the unfolded protein response. Antioxid Redox Signal.
9:2357–2371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Li Y, Guo Y, Tang J, Jiang J and Chen Z:
New insights into the roles of CHOP-induced apoptosis in ER stress.
Acta Biochim Biophys Sin (Shanghai). 46:629–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Palam LR, Baird TD and Wek RC:
Phosphorylation of eIF2 facilitates ribosomal bypass of an
inhibitory upstream ORF to enhance CHOP translation. J Biol Chem.
286:10939–10949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alplha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang J, Kang R, Huang H, Xi X, Wang B,
Wang J and Zhao Z: Hepatitis C virus core protein activates
autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B
and ATG12 expression. Autophagy. 10:766–784. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ma X, Piao S, Dey S, Mcafee Q, Karakousis
G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al: Targeting
ER stress-induced autophagy overcomes BRAF inhibitor resistance in
melanoma. J Clin Invest. 124:1406–1417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rzymski T, Milani M, Pike L, Buffa F,
Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I and Harris
AL: Regulation of autophagy by ATF4 in response to severe hypoxia.
Oncogene. 29:4424–4435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
B'Chir W, Chaveroux C, Carraro V, Averous
J, Maurin AC, Jousse C, Muranishi Y, Parry L, Fafournoux P and
Bruhat A: Dual role for CHOP in the crosstalk between autophagy and
apoptosis to determine cell fate in response to amino acid
deprivation. Cell Signal. 26:1385–1391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Li Y, Guo Y, Tang J, Jiang J and Chen Z:
New insights into the roles of CHOP-induced apoptosis in ER stress.
Acta Biochim Biophys Sin (Shanghai). 46:629–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Liu K, Shi Y, Guo X, Wang S, Ouyang Y, Hao
M, Liu D, Qiao L, Li N, Zheng J and Chen D: CHOP mediates
ASPP2-induced autophagic apoptosis in hepatoma cells by releasing
Beclin-1 from Bcl-2 and inducing nuclear translocation of Bcl-2.
Cell Death Dis. 5:e13232014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rouschop KM, van den Beucken T, Dubois L,
Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W,
Voncken JW, et al: The unfolded protein response protects human
tumor cells during hypoxia through regulation of the autophagy
genes MAP1LC3B and ATG5. J Clin Invest. 120:127–141. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Puthalakath H, O'Reilly LA, Gunn P, Lee L,
Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin
J, Motoyama N, et al: ER stress triggers apoptosis by activating
BH3-only protein Bim. Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gorman AM, Healy SJ, Jager R and Samali A:
Stress management at the ER: Regulators of ER stress-induced
apoptosis. Pharmacol Ther. 134:306–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ohoka N, Yoshii S, Hattori T, Onozaki K
and Hayashi H: TRB3, a novel ER stress-inducible gene, is induced
via ATF4-CHOP pathway and is involved in cell death. EMBO J.
24:1243–1255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Du K, Herzig S, Kulkarni RN and Montminy
M: TRB3: A tribbles homolog that inhibits Akt/PKB activation by
insulin in liver. Science. 300:1574–1577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
B'Chir W, Maurin AC, Carraro V, Averous J,
Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P and Bruhat
A: The eIF2α/ATF4 pathway is essential for stress-induced autophagy
gene expression. Nucleic Acids Res. 41:7683–7699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Shen J, Chen X, Hendershot L and Prywes R:
ER stress regulation of ATF6 localization by dissociation of
BiP/GRP78 binding and unmasking of Golgi localization signals. Dev
Cell. 3:99–111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ye J, Rawson RB, Komuro R, Chen X, Davé
UP, Prywes R, Brown MS and Goldstein JL: ER stress induces cleavage
of membrane-bound ATF6 by the same proteases that process SREBPs.
Mol Cell. 6:1355–1364. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bommiasamy H, Back SH, Fagone P, Lee K,
Meshinchi S, Vink E, Sriburi R, Frank M, Jackowski S, Kaufman RJ
and Brewer JW: ATF6alpha induces XBP1-independent expansion of the
endoplasmic reticulum. J Cell Sci. 122:1626–1636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Adachi Y, Yamamoto K, Okada T, Yoshida H,
Harada A and Mori K: ATF6 is a transcription factor specializing in
the regulation of quality control proteins in the endoplasmic
reticulum. Cell Struct Funct. 33:75–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gade P, Ramachandran G, Maachani UB, Rizzo
MA, Okada T, Prywes R, Cross AS, Mori K and Kalvakolanu DV: An
IFN-γ-stimulated ATF6-C/EBP-β-signaling pathway critical for the
expression of Death Associated Protein Kinase 1 and induction of
autophagy. Proc Natl Acad Sci USA. 109:10316–10321. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zalckvar E, Berissi H, Mizrachy L,
Idelchuk Y, Koren I, Eisenstein M, Sabanay H, PinkasKramarski R and
Kimchi A: DAP-kinase-mediated phosphorylation on the BH3 domain of
beclin 1 promotes dissociation of beclin 1 from Bcl-XL and
induction of autophagy. EMBO Rep. 10:285–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cook KL, Shajahan AN, Wärri A, Jin L,
HilakiviClarke LA and Clarke R: Glucose-regulated protein 78
controls cross-talk between apoptosis and autophagy to determine
antiestrogen responsiveness. Cancer Res. 72:3337–3349. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Shimada Y, Kobayashi H, Kawagoe S, Aoki K,
Kaneshiro E, Shimizu H, Eto Y, Ida H and Ohashi T: Endoplasmic
reticulum stress induces autophagy through activation of p38 MAPK
in fibroblasts from Pompe disease patients carrying c.546G>T
mutation. Mol Genet Metab. 104:566–573. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kim DS, Kim JH, Lee GH, Kim HT, Lim JM,
Chae SW, Chae HJ and Kim HR: p38 Mitogen-activated protein kinase
is involved in endoplasmic reticulum stress-induced cell death and
autophagy in human gingival fibroblasts. Biol Pharm Bull.
33:545–549. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Carloni S, Albertini MC, Galluzzi L,
Buonocore G, Proietti F and Balduini W: Increased autophagy reduces
endoplasmic reticulum stress after neonatal hypoxia-ischemia: Role
of protein synthesis and autophagic pathways. Exp Neurol.
255:103–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Verchot J: The ER quality control and ER
associated degradation machineries are vital for viral
pathogenesis. Front Plant Sci. 5:662014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential Effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
JoshiBarr S, Bett C, Chiang WC, Trejo M,
Goebel HH, Sikorska B, Liberski P, Raeber A, Lin JH, Masliah E and
Sigurdson CJ: De novo prion aggregates trigger autophagy in
skeletal muscle. J Virol. 88:2071–2082. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bernales S, McDonald KL and Walter P:
Autophagy counterbalances endoplasmic reticulum expansion during
the unfolded protein response. PLoS Biol. 4:e4232006. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Vincenz L, Jäger R, O'Dwyer M and Samali
A: Endoplasmic reticulum stress and the unfolded protein response:
Targeting the Achilles heel of multiple myeloma. Mol Cancer Ther.
12:831–843. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L and Tang D: HMGB1 promotes drug resistance in
osteosarcoma. Cancer Res. 72:230–238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Huang J, Liu K, Yu Y, Xie M, Kang R,
Vernon P, Cao L, Tang D and Ni J: Targeting HMGB1-mediated
autophagy as a novel therapeutic strategy for osteosarcoma.
Autophagy. 8:275–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yang L, Yang S, Liu J, Wang X, Ji J, Cao
Y, Lu K, Wang J and Gao Y: Expression of GRP78 predicts
taxane-based therapeutic resistance and recurrence of human gastric
cancer. Exp Mol Pathol. 96:235–241. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Bifulco G, Miele C, Di Jeso B, Beguinot F,
Nappi C, Di Carlo C, Capuozzo S, Terrazzano G, Insabato L and
Ulianich L: Endoplasmic reticulum stress is activated in
endometrial adenocarcinoma. Gynecol Oncol. 125:220–225. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Visioli F, Wang Y, Alam GN, Ning Y, Rados
PV, Nör JE and Polverini PJ: Glucose-regulated protein 78 (Grp78)
confers chemoresistance to tumor endothelial cells under acidic
stress. PLoS ONE. 9:e1010532014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lin JA, Fang SU, Su CL, Hsiao CJ, Chang
CC, Lin YF and Cheng CW: Silencing glucose-regulated protein 78
induced renal cell carcinoma cell line G1 cell-cycle arrest and
resistance to conventional chemotherapy. Urol Oncol.
32:29.e1–29.e11. 2014. View Article : Google Scholar
|
|
102
|
KosakowskaCholody T, Lin J, Srideshikan
SM, Scheffer L, Tarasova NI and Acharya JK: HKH40A downregulates
GRP78/BiP expression in cancer cells. Cell Death Dis. 5:e12402014.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Reddy RK, Mao C, Baumeister P, Austin RC,
Kaufman RJ and Lee AS: Endoplasmic reticulum chaperone protein
GRP78 protects cells from apoptosis induced by topoisomerase
inhibitors: Role of ATP binding site in suppression of caspase-7
activation. J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Fu Y, Li J and Lee AS: GRP78/BiP inhibits
endoplasmic reticulum BIK and protects human breast cancer cells
against estrogen starvation-induced apoptosis. Cancer Res.
67:3734–3740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhou H, Zhang Y, Fu Y, Chan L and Lee AS:
Novel mechanism of anti-apoptotic function of 78-kDa
glucose-regulated protein (GRP78): Endocrine resistance factor in
breast cancer, through release of B-cell lymphoma 2 (BCL-2) from
BCL-2-interacting killer (BIK). J Biol Chem. 286:25687–25696. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shani G, Fischer WH, Justice NJ, Kelber
JA, Vale W and Gray PC: GRP78 and Cripto form a complex at the cell
surface and collaborate to inhibit transforming growth factor beta
signaling and enhance cell growth. Mol Cell Biol. 28:666–677. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lee E, Nichols P, Spicer D, Groshen S, Yu
MC and Lee AS: GRP78 as a novel predictor of responsiveness to
chemotherapy in breast cancer. Cancer Res. 66:7849–7853. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Suyama K, Watanabe M, Sakabe K, Okada Y,
Matsuyama D, Kuroiwa M and Mochida J: Overexpression of GRP78
protects glial cells from endoplasmic reticulum stress. Neurosci
Lett. 504:271–276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Li Z, Wang Y, Wu H, Zhang L, Yang P and Li
Z: GRP78 enhances the glutamine metabolism to support cell survival
from glucose deficiency by modulating the β-catenin signaling.
Oncotarget. 5:5369–5380. 2014.PubMed/NCBI
|
|
112
|
Li W, Wang W, Dong H, Li Y, Li L, Han L,
Han Z, Wang S, Ma D and Wang H: Cisplatin-induced senescence in
ovarian cancer cells is mediated by GRP78. Oncol Rep. 31:2525–2534.
2014.PubMed/NCBI
|
|
113
|
Auf G, Jabouille A, Delugin M, Guerit S,
Pineau R, North S, Platonova N, Maitre M, Favereaux A, Vajkoczy P,
et al: High epiregulin expression in human U87 glioma cells relies
on IRE1α and promotes autocrine growth through EGF receptor. BMC
Cancer. 13:5972013. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Auf G, Jabouille A, Guérit S, Pineau R,
Delugin M, Bouchecareilh M, Magnin N, Favereaux A, Maitre M, Gaiser
T, et al: Inositol-requiring enzyme 1alpha is a key regulator of
angiogenesis and invasion in malignant glioma. Proc Natl Acad Sci U
S A. 107:15553–15558. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Tay KH, Luan Q, Croft A, Jiang CC, Jin L,
Zhang XD and Tseng HY: Sustained IRE1 and ATF6 signaling is
important for survival of melanoma cells undergoing ER stress. Cell
Signal. 26:287–294. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Thorpe JA and Schwarze SR: IRE1alpha
controls cyclin A1 expression and promotes cell proliferation
through XBP-1. Cell Stress Chaperones. 15:497–508. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Gambella M, Rocci A, Passera R, Gay F,
Omedè P, Crippa C, Corradini P, Romano A, Rossi D, Ladetto M, et
al: High XBP1 expression is a marker of better outcome in multiple
myeloma patients treated with bortezomib. Haematologica.
99:e14–e16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bagratuni T, Wu P, Gonzalez de Castro D,
Davenport EL, Dickens NJ, Walker BA, Boyd K, Johnson DC, Gregory W,
Morgan GJ and Davies FE: XBP1s levels are implicated in the biology
and outcome of myeloma mediating different clinical outcomes to
thalidomide-based treatments. Blood. 116:250–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chen X, Iliopoulos D, Zhang Q, Tang Q,
Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et
al: XBP1 promotes triple-negative breast cancer by controlling the
HIF1α pathway. Nature. 508:103–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Mimura N, Fulciniti M, Gorgun G, Tai YT,
Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, et al:
Blockade of XBP1 splicing by inhibition of IRE1α is a promising
therapeutic option in multiple myeloma. Blood. 119:5772–5781. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Schroder M and Kaufman RJ: Divergent roles
of IRE1alpha and PERK in the unfolded protein response. Curr Mol
Med. 6:5–36. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Cullinan SB and Diehl JA: PERK-dependent
activation of Nrf2 contributes to redox homeostasis and cell
survival following endoplasmic reticulum stress. J Biol Chem.
279:20108–20117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
BobrovnikovaMarjon E, Grigoriadou C, Pytel
D, Zhang F, Ye J, Koumenis C, Cavener D and Diehl JA: PERK promotes
cancer cell proliferation and tumor growth by limiting oxidative
DNA damage. Oncogene. 29:3881–3895. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Krishnamoorthy J, Rajesh K, Mirzajani F,
Kesoglidou P, Papadakis AI and Koromilas AE: Evidence for eIF2α
phosphorylation-independent effects of GSK2656157, a novel
catalytic inhibitor of PERK with clinical implications. Cell Cycle.
13:801–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Milani M, Rzymski T, Mellor HR, Pike L,
Bottini A, Generali D and Harris AL: The role of ATF4 stabilization
and autophagy in resistance of breast cancer cells treated with
bortezomib. Cancer Research. 69:4415–4423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Hamanaka RB, BobrovnikovaMarjon E, Ji X,
Liebhaber SA and Diehl JA: PERK-dependent regulation of IAP
translation during ER stress. Oncogene. 28:910–920. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
KusioKobialka M, PodszywalowBartnicka P,
Peidis P, GlodkowskaMrowka E, Wolanin K, Leszak G, Seferynska I,
Stoklosa T, Koromilas AE and Piwocka K: The PERK-eIF2α
phosphorylation arm is a pro-survival pathway of BCR-ABL signaling
and confers resistance to imatinib treatment in chronic myeloid
leukemia cells. Cell Cycle. 11:4069–4078. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Higa A, Taouji S, Lhomond S, Jensen D,
FernandezZapico ME, Simpson JC, Pasquet JM, Schekman R and Chevet
E: Endoplasmic reticulum stress-activated transcription factor
ATF6α requires the disulfide isomerase PDIA5 to modulate
chemoresistance. Mol Cell Biol. 34:1839–1849. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Schewe DM and Aguirre-Ghiso JA:
ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor
cells in vivo. Proc Natl Acad Sci U S A. 105:10519–10524. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Shimodaira Y, Takahashi S, Kinouchi Y,
Endo K, Shiga H, Kakuta Y, Kuroha M and Shimosegawa T: Modulation
of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP
homologous protein (CHOP) and inositol-requiring enzyme 1α (IRE1α)
in human colon cancer cells. Biochem Biophys Res Commun.
445:524–533. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Mahoney E, Lucas DM, Gupta SV, Wagner AJ,
Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, et
al: ER stress and autophagy: New discoveries in the mechanism of
action and drug resistance of the cyclin-dependent kinase inhibitor
flavopiridol. Blood. 120:1262–1273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Dey S, Tameire F and Koumenis C: PERK-ing
up autophagy during MYC-induced tumorigenesis. Autophagy.
9:612–614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ma X, Piao S, Dey S, Mcafee Q, Karakousis
G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al: Targeting
ER stress-induced autophagy overcomes BRAF inhibitor resistance in
melanoma. J Clin Invest. 124:1406–1417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Hart LS, Cunningham JT, Datta T, Dey S,
Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al: ER
stress-mediated autophagy promotes Myc-dependent transformation and
tumor growth. J Clin Invest. 122:4621–4634. 2012. View Article : Google Scholar : PubMed/NCBI
|