Introduction
Hepatocellular carcinoma (HCC) is one of the most
frequent tumors of the liver, and it is reported to account for 5%
of all malignant neoplasms (1,2). The
aggressiveness and wide dissemination of HCC frequently leads to
mortality in the affected population (3), and despite the development of modern
treatment protocols, the incidence and mortality rates of the
disease remain high (3). Therefore,
investigation into novel methods to improve the treatment and
survival of liver cancer patients is essential. The pathogenesis of
liver cancer is complex, and studies have demonstrated that
uncontrolled proliferation, various ion disorders and resistance to
apoptosis are key features of this process (4). However, the exact mechanisms of
pathogenesis are unclear, particularly that of apoptosis
resistance.
It has been demonstrated that apoptosis, a type of
programmed cell death, is vital in the prevention of HCC
development (5). The activation of
apoptotic pathways is a key mechanism by which HCC cells may be
killed, and defects in apoptotic signaling can lead to the drug
resistance of these cells (6).
Therefore, the induction of apoptosis is considered to be an
important method in the assessment of the clinical effectiveness of
anti-HCC drugs.
Notably, the phosphoinositide 3-kinase/protein
kinase B (PI3K/Akt) pathway, which has been demonstrated to play a
critical role in regulating cell growth and cell survival in
different systems, has been identified to be involved in the
pathogenesis of HCC by inducing the apoptosis resistance of HCC
cells (7). The PI3K kinase is
composed of a catalytic subunit, p110, and a regulatory subunit,
p85; the activation of PI3K depends upon the activation of the p85
subunit. When p85 is activated, it directs signals to phosphorylate
Akt, which subsequently provides signals that regulate apoptosis
resistance (8). Inhibiting PI3K/Akt
signaling has been reported to induce apoptosis in HCC cells
(9,10).
For the past several decades, rosiglitazone (RGZ),
an agonist of peroxisome proliferator-activated receptor γ
(PPAR-γ), has been extensively used in clinical practice due to its
important regulatory role in energy homeostasis, and lipid and
glucose metabolism (11). PPAR-γ is
widely distributed in HCC cells (12). It has been demonstrated that RGZ
regulates the activity of transcription factors essential for
apoptosis, and it has also been used to induce apoptosis in
leukemia cells and lung cancer cells though the PI3K-Akt signaling
pathway (13,14). However, the effect of RGZ on HCC cells
is yet to be elucidated. Therefore, the present study investigated
its effects in the classical human HCC HepG2 cell line (15) to address this question.
Materials and methods
Main reagents
RGZ, purchased from Cayman Chemical Company (Ann
Arbor, MI, USA), was dissolved in dimethyl sulfoxide (DMSO) and
stored at −20°C. GW9662, a PPAR-γ antagonist, was also purchased
from Cayman Chemical Company. The Annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI) Apoptosis Detection Kit
was purchased from R&D Systems, Inc. (Minneapolis, MN,
USA).
Cell culture
The human HCC HepG2 cell line was obtained from the
American Type Culture Collection (Rockville, MD, USA). The cells
were plated at a density of 2×105 cells/cm2
and cultured in Dulbecco's modified Eagle's medium containing 10%
fetal bovine serum (Gibco-BRL Life Technologies, Grand Island, NY,
USA) and penicillin/streptomycin (100 U/l; Wuhan Goodbio Technology
Co. Ltd., Wuhan, China), at 37°C in a humidified atmosphere of 5%
CO2 and 95% air.
Cell viability rate
The cell viability rate was assessed using the
microculture tetrazolium method. The cells were plated at a density
of 3×104/cm2 in 96-well plates. The HepG2
cells contained 6 parallel wells were divided into the control, RGZ
and GW9662 groups. They were incubated with DMSO, RGZ and
RGZ+GW9662 for 72 h, respectively, and 10 µl MTT working solution
was added, followed by continuous incubation for 4 h. All culture
medium supernatant was removed from each well following
centrifugation at 3,000 × g, and replaced with 100 µl DMSO.
Following thorough solubilization, the absorbance (A) of each well
was measured using a microculture plate reader at 570 nm. The cell
inhibitory rate was calculated according to the following formula:
Inhibitory rate = 100 × (Acontrol group - Atreated
group) / Acontrol group).
Flow cytometric (FCM) analysis of cell
apoptosis
Apoptosis was assessed using an Annexin V-FITC/PI
Apoptosis Detection Kit. For FCM analysis, 2×105
cells/well were treated with different concentrations of RGZ (0,
20, 40, 80 µg/ml) or GW9662 (0, 5, 10, 20 µg/ml). The cells were
subsequently collected, pelleted and washed with phosphate-buffered
saline (PBS) prior to fixing overnight at −20°C in 75% ethanol. The
cells were washed again with PBS, resuspended and treated with
RNase (200 mg/l) for 30 min at 37°C, prior to incubation with 20
mg/l PI in the dark for 15 min. The suspension was passed through a
nylon mesh filter and underwent FCM analysis (FACSort™;
Becton-Dickinson, Franklin Lakes, NJ, USA). All data were
collected, stored and analyzed by LYSIS II software
(Becton-Dickinson). The experiments were repeated three times, and
the results are presented as the mean ± standard deviation
(SD).
Western blotting
Total proteins from HepG2 cells in the three groups
were separated by 12% SDS-PAGE (Wuhan Goodbio Technology Co. Ltd.),
and the separated proteins were electrotransferred onto
nitrocellulose membranes (Wuhan Goodbio Technology Co. Ltd.) using
a Trans-Blot® Turbo™ Transfer System (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The membranes were first blocked with 5%
non-fat milk for 2 h at room temperature, prior to incubation with
the following primary monoclonal anti-human antibodies: PPAR-γ,
activated PPAR-γ (p-PPAR-γ; rabbit; 1:500; cat. no. ab195925;
Abcam, Cambridge, UK), PPAR-γ (1:1,000; rabbit; cat. no. ab59256;
Abcam) p85, p-p85, Akt, p-Akt, caspase 3 (rabbit; 1:500; cat. no.
ab32351; Abcam), cleavage-caspase 3 (rabbit; 1:500; cat. no.
ab2302; Abcam), Bax (rabbit; 1:1,000; cat. no. ab7977; Abcam),
Bcl-2 (mouse; 1:500; cat. no. ab117115; Abcam), p-p85 (rabbit;
1:1,000; cat. no. 4228S; Cell Signaling Technology, Inc., Danvers,
MA, USA), p85 (rabbit; 1:500; cat. no. 4292S; Cell Signaling
Technology, Inc.), p-Akt (rabbit; 1:500; cat. no. 4060S; Cell
Signaling Technology, Inc.), Akt (rabbit; 1:1,000; cat. no. 4685S;
Cell Signaling Technology, Inc.) and β-actin (rabbit; 1:3,000; cat.
no. ab6276; Abcam). Anti-rabbit antibody conjugated to horseradish
peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA,
USA) was used as the secondary antibody. β-actin was used as an
intrinsic quality control. The bands were incubated in ECL Plus
reagent (Amersham, Piscataway, NJ, USA) and chemiluminescence was
detected on BioMax MR Film (Kodak, Rochester, NY, USA). The density
of the bands was quantified using Labworks image acquisition and
analysis software (UVP LLC, Upland, CA, USA) (16).
Statistical analysis
All experiments were performed in triplicate, and
the results are expressed as the mean ± SD. For the statistical
analysis, Student's t-tests were performed using SPSS software,
version 12.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
RGZ significantly inhibits the cell
viability of HepG2 cells
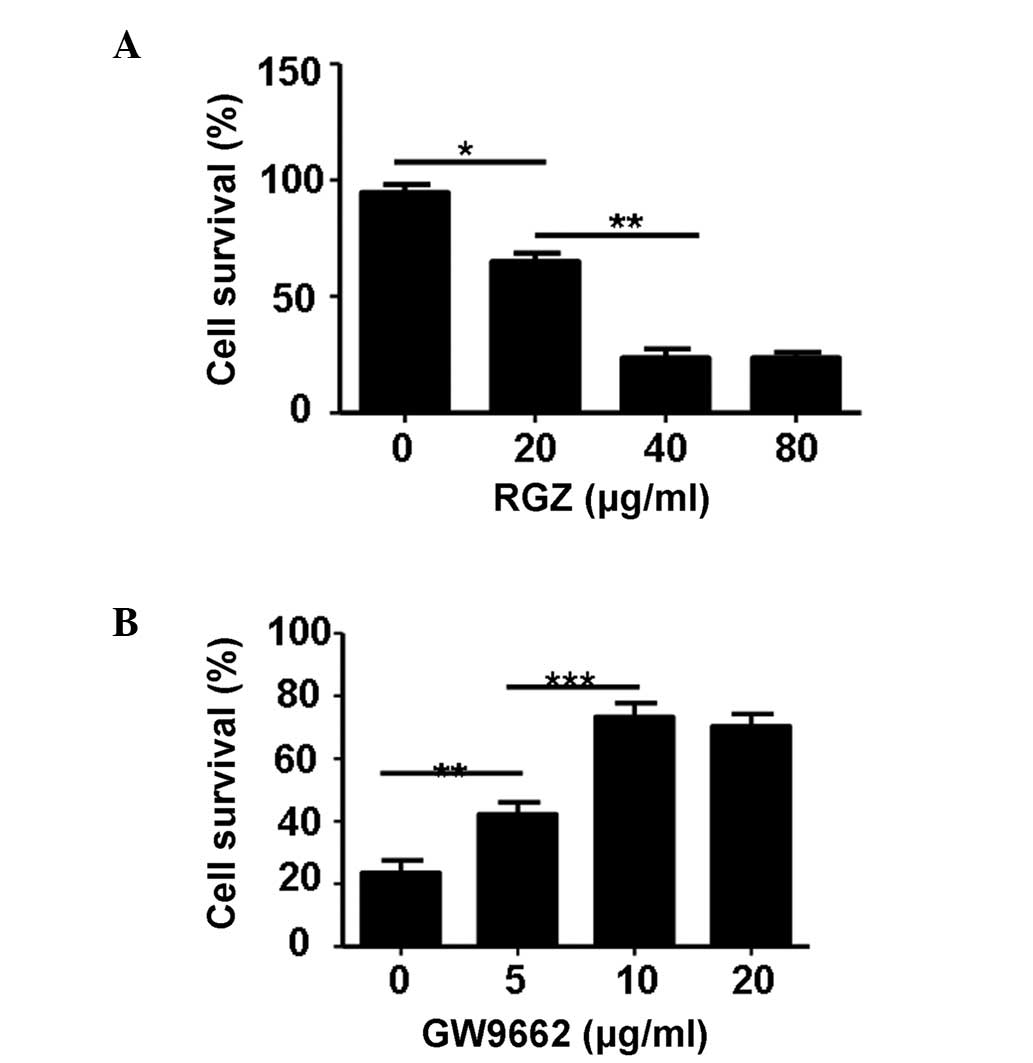
The cytotoxic effect of RGZ on HepG2 cells was
determined following incubation with varying concentrations of RGZ
by MTT assay. As shown in Fig. 1A,
RGZ treatment significantly attenuated the cell viability of HepG2
cells, with a concentration of 40 µg/ml at 72 h producing the
optimal effect (P<0.01). Inhibition of cell viability by RGZ was
dose-dependent from 0–40 µg/ml. In order to further demonstrate
that the cytotoxic effect on HepG2 cell viability was caused by
RGZ, the cells were treated with various concentrations (0, 5, 10
and 20 µg/ml) of PPAR-γ antagonist, GW9662, plus RGZ (40 µg/ml). As
shown in Fig. 1B, GW9662
significantly attenuated the cytotoxic effect of RGZ in the HepG2
cells. The optimal concentration of GW9662 to attenuate the
cytotoxic effect of RGZ was 10 µg/ml (P<0.001).
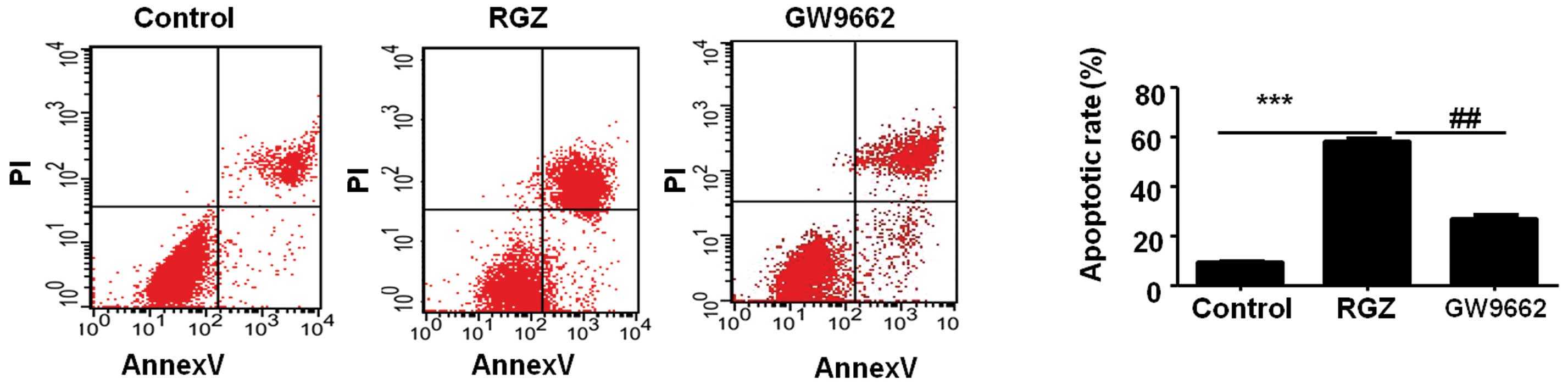
RGZ induces the apoptosis of HepG2
cells
The most effective concentrations of RGZ and GW9662
were used to further analyze the effect of RGZ on the HepG2 cell
lines. The effect of RGZ on the apoptosis of the HepG2 cell lines
was examined by FCM analysis. Compared with the HepG2 cells from
the control group, the RGZ-treated cells exhibited a higher rate of
apoptosis (P<0.001). Notably, the HepG2 cells in the
GW9662-treated group exhibited a 1.3-fold lower rate of apoptosis
compared with the cells in the RGZ-treated group (P<0.01). These
results indicated that the administration of RGZ may significantly
induce apoptosis in the HepG2 cells (Fig.
2).
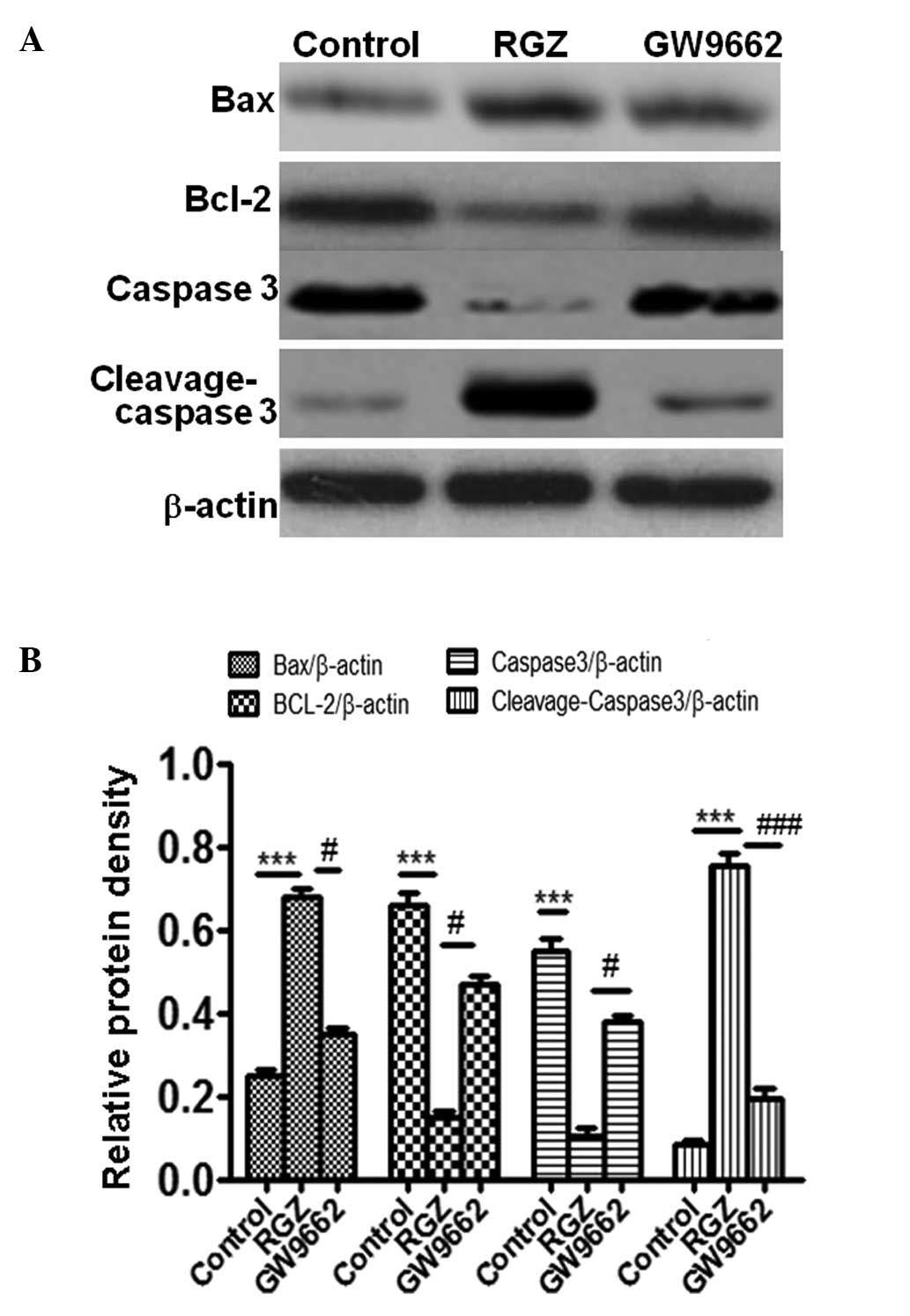
It has been previously demonstrated than Bax/Bcl-2
protein are associated with apoptosis (17,18). In
order to further demonstrate the effect of RGZ on apoptosis, the
expression of Bax and Bcl-2 was examined by western blotting in
RGZ-treated HepG2 cells. As shown in Fig.
3, RGZ-treated cells exhibited increased expression of Bax and
reduced expression of Bcl-2 compared with the cells in the control
(P<0.001) and GW9662-treated (P<0.05) groups. This result was
a further indication that RGZ was able to induce apoptosis in the
HepG2 cells.
Caspase 3 activation has previously been
demonstrated to serve an important role in apoptosis (17,18). The
present results demonstrated that administration of RGZ can
significantly reduce levels of caspase 3 (P<0.001 and P<0.05
compared with the control and GW9662-treated cells, respectively)
and increase cleavage-caspase 3 (P<0.001 compared with the which
further indicated that RGZ could induce the apoptosis of HepG2
cells (P<0.05).
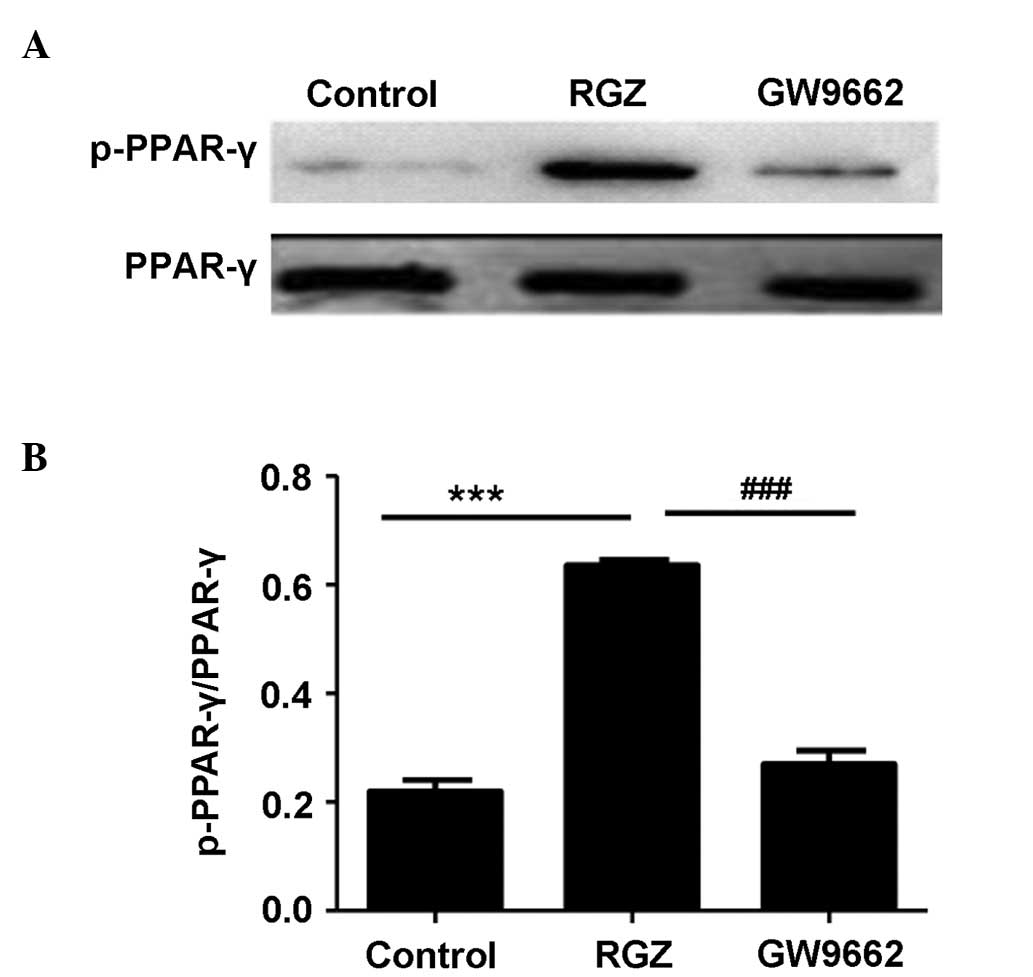
RGZ induces apoptosis through PPAR-γ
activation
As the first step to addressing the underlying
mechanisms of the RGZ-induced apoptosis of HepG2 cells, PPAR-γ
activation was examined. RGZ is an agonist for PPAR-γ, while GW9662
is an antagonist. Unexpectedly, RGZ and GW9662 administration
exerted no perceptible effect on PPAR-γ expression (P>0.05).
However, the amount of activated p-PPAR-γ observed in the HepG2
cells following RGZ administration was significantly higher
compared with that of the control (P<0.001) and GW9662-treated
(P<0.01) cells. These results indicated that RGZ was able to
induce PPAR-γ activation, while GW9662 suppressed the effect of RGZ
on PPAR-γ activation (Fig. 4).
PPAR-γ activation downregulates
PI3K/Akt signaling
The aforementioned results prompted the
investigation of the impact of PPAR-γ activation on PI3K/Akt
signaling, a well-established pathway that has significant
implications in HepG2 cells (15). To
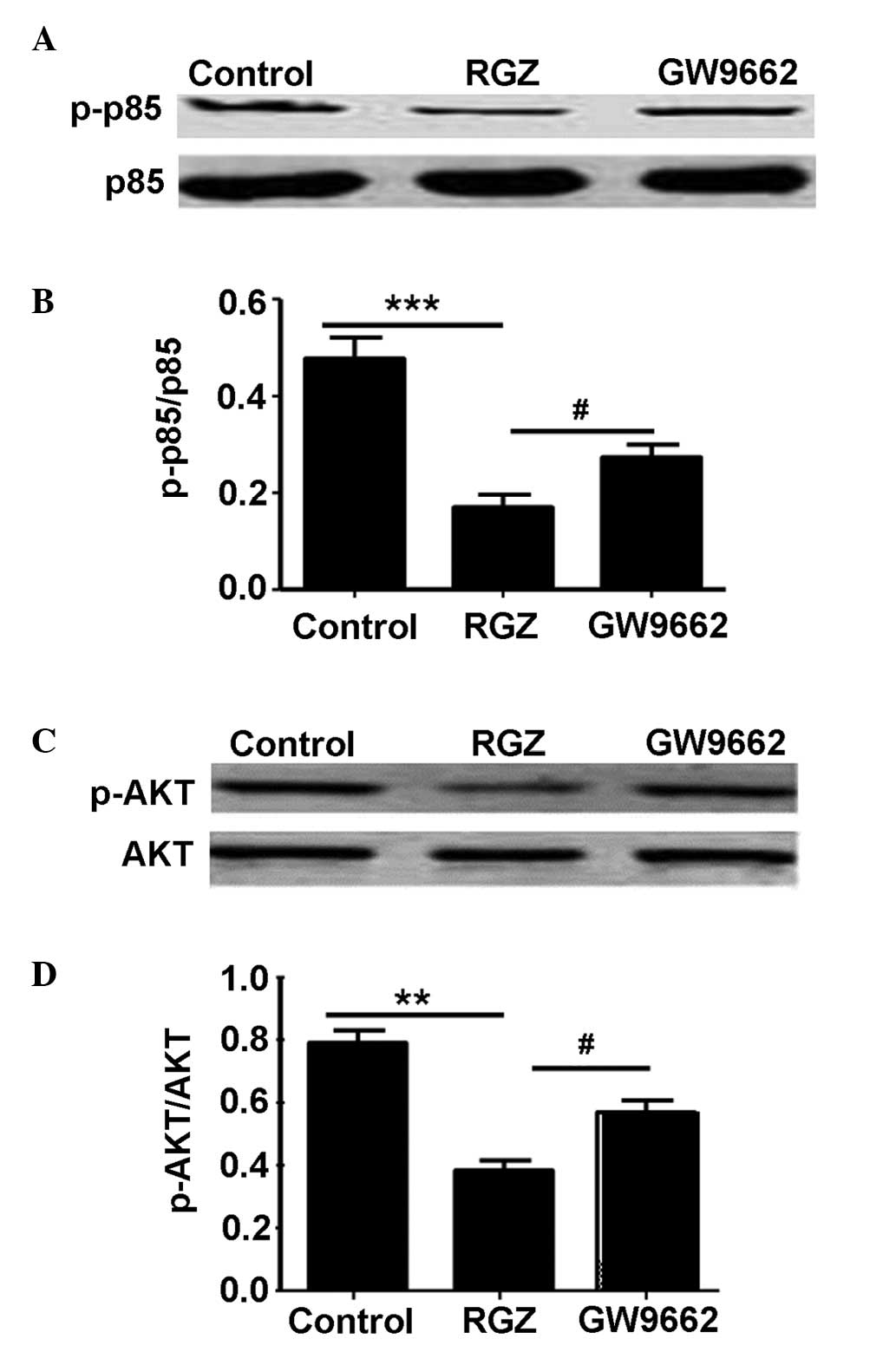
this end, the activity of the PI3K p85 regulatory subunit was first
examined. No significant difference in total p85 levels was
detected between the 3 groups of HepG2 cells (P>0.05; Fig. 5A). However, markedly lower levels of
p-p85 were noted in the RGZ-treated group compared with the
GW9662-treated or control groups (P<0.05 and P<0.001,
raspectively; Fig. 5B). As p85
activation provides signals to Akt, Akt activity was subsequently
examined. Similar to p85, no difference was observed in total Akt
levels between the groups (P>0.05; Fig. 5C), whilst p-Akt levels were
significantly lower in the RGZ-treated group compared with the
control (P<0.01) or GW9662-treated (P<0.05) groups.
Collectively, these data suggested that RGZ treatment increased
PPAR-γ activation, and that p-PPAR-γ attenuated PI3K p85 activity,
which subsequently decreased Akt activation (Fig. 5D).
Discussion
Although RGZ has been used extensively in clinical
practice in the treatment of diabetes (11), its impact on HCC cells remains to be
studied. Therefore, its effects on HepG2 cells were investigated in
the present study. RGZ treatment was found to significantly
attenuate cell viability and induce apoptosis in HepG2 cells. The
effect of RGZ on cell viability was partially dose-dependent, and a
concentration of 40 µg/ml was found to produce the greatest effect.
GW9662, an antagonist of PPAR-γ, was observed to suppress the
effect of RGZ on cell viability in HepG2 cells in a partially
dose-dependent manner, with an optimal concentration of 10 µg/ml.
Western blot analysis revealed that RGZ treatment increased the
expression of Bax and cleavage-caspase 3 and reduced the expression
Bcl-2 and caspase 3 proteins. FCM demonstrated that RGZ could
increase the apoptosis rate of the HepG2 cells, with a
concentration of 40 µg/ml producing the greatest effect. GW9662
suppressed the effects of RGZ on the Bax, cleavage-caspase 3, Bcl-2
and caspase 3 protein expression in the HepG2 cells. These results
are consistent with the hypothesis that RGZ is able to induce
apoptosis in HepG2 cells. In concordance with these results, a
number of previous studies have reported that RGZ induces apoptosis
in leukemia K562 and cholangiocarcinoma QBC939 cells (19,20).
Together, these results suggest that RGZ may be a novel therapeutic
agent for the treatment of liver cancer in the clinical setting.
However, there is a lack of evidence with regard to the effect of
RGZ treatment on HCC in vivo. The anti-HCC effects of RGZ
therefore require further investigation.
To ascertain the molecular mechanisms by which RGZ
induces the apoptosis of HepG2 cells, its effect on PPAR-γ
activation were examined. Unexpectedly, RGZ and GW9662 treatment
did not affect PPAR-γ expression. However, administration of RGZ
was observed to upregulate p-PPAR-γ expression, while GW9662
reduced the expression of p-PPAR-γ. This may be as RGZ activates
PPAR-γ, rather than increasing its expression, leading to the
apoptosis of the HepG2 cells.
A number of studies have demonstrated that PPAR-γ
activation has a therapeutic effect on cancer cells that depends on
the induction of apoptosis (20).
Therefore, pathways downstream of PPAR-γ associated with the
regulation of apoptosis, including PI3K/Akt signaling, were
examined next in the present study. It has been demonstrated
PI3K/Akt signaling is important in inducing anti-apoptotic effects
in chronic myeloid leukemia (21) and
that PPAR-γ activation can regulate PI3K/Akt signaling (22). In the present study, PPAR-γ activation
had no effect on the expression of the PI3K regulatory subunit,
p85, however, it did attenuate p85 activation, as shown by the
significantly lower levels of p-p85 in the RGZ-treated HepG2 cells
compared with the control cells. This result prompted the
investigation of Akt activity, as p-p85 leads to Akt activation. In
line with the aforementioned results, RGZ significantly decreased
the Akt activity, as indicated by the downregulation of p-Akt with
no effect on the expression of Akt. Taken together, these data
suggest that RGZ may treat liver cancer cells by enhancing PPAR-γ
activation, through which PI3K/Akt signaling activation is
suppressed, thus inducing apoptosis.
The PI3K/Akt pathway has long been recognized to be
important in regulating the immune response (23,24).
Different PI3K heterodimers control cell survival, proliferation,
B- and T-cell receptor signaling and chemotaxis in B and T
lymphocytes (23,24). More recently, it has been reported
that the PI3K/Akt pathway has versatile roles in apoptosis in
various cell types, including K562 cells, lung cancer cells,
monocytes, macrophages and parenchymal cells (25,26).
Therefore, the present study conducted an additional investigation
into the effect that blocking PI3K/Akt signaling had on apoptosis
in the HepG2 cells. Given the capacity of RGZ treatment to induce
apoptosis, it is worth noting that PPAR-γ activation induced by RGZ
may be involved in additional pathways other than the PI3K/Akt
signaling, such as the MAPK kinase cascade (27). As a result, further studies focusing
on the pathways associated with PPAR-γ activation during the
induction of HepG2 cell apoptosis are necessary.
In summary, the current study presents evidence that
RGZ affects the induction of apoptosis in HepG2 cells in
vitro, and that the mechanism involves the stimulation of
PPAR-γ to suppress PI3K/Akt signaling activation. Therefore, RGZ
may be a promising therapy for the treatment of liver cancer in the
clinical setting.
Acknowledgements
The authors would like to thank the members of the
Central Laboratories of Weifang Medical College (Weifang, China)
for their insight and technical support. This study was supported
by grants from the National Natural Foundation of China (grant no.
81100264).
References
|
1
|
Alves RC, Alves D, Guz B, Matos C, Viana
M, Harriz M, et al: Advanced hepatocellular carcinoma. Review of
targeted molecular drugs. Ann Hepatol. 10:21–27. 2011.PubMed/NCBI
|
|
2
|
WalyRaphael S, Yangde Z and Yuxiang C:
Hepatocellular carcinoma: focus on different aspects of management.
ISRN Oncol. 2012:4216732012.PubMed/NCBI
|
|
3
|
Johnson PJ, Qin S, Park JW, Poon RT, Raoul
JL, Philip PA, et al: Brivanib versus sorafenib as first-line
therapy in patients with unresectable, advanced hepatocellular
carcinoma: results from the randomized phase III BRISK-FL study. J
Clin Oncol. 31:3517–3524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blackadar CB: Systematic review of
hepatocellular carcinoma mortality rates among hepatitis B
virus-infected renal transplant recipients, with supplemental
analyses of liver failure and all-cause mortality. Int J Infect
Dis. 17:e24–e36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin PH, Liu X, Qiu YY, Cai JF, Qin JM, Zhu
HR and Li Q: Anti-tumor activity and apoptosis-regulation
mechanisms of bufalin in various cancers: new hope for cancer
patients. Asian Pac J Cancer Prev. 13:5339–5343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamaguchi M: Suppressive role of
regucalcin in liver cell proliferation: involvement in
carcinogenesis. Cell Prolif. 46:243–253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fabregat I: Dysregulation of apoptosis in
hepatocellular carcinoma cells. World J Gastroenterol. 15:513–520.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ehrhardt C and Ludwig S: A new player in a
deadly game: influenza viruses and the PI3K/Akt signalling pathway.
Cell Microbiol. 11:863–871. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang CZ, Wang XD, Wang HW, Cai Y and Chao
LQ: Sorafenib inhibits liver cancer growth by decreasing mTOR, AKT,
and PI3K expression. J BUON. 20:218–222. 2015.PubMed/NCBI
|
|
10
|
Yan CM, Chai EQ, Cai HY, Miao GY and Ma W:
Oleuropein induces apoptosis via activation of caspases and
suppression of phosphatidylinositol 3-kinase/protein kinase B
pathway in HepG2 human hepatoma cell line. Mol Med Rep.
11:4617–4624. 2015.PubMed/NCBI
|
|
11
|
Beliaeva MI: Modern views of causes of
microvascular complications development and progression in type 2
diabetes mellitus and peculiarities of their treatment. Vestn
Oftalmol. 129:70–75. 2013.(In Russian). PubMed/NCBI
|
|
12
|
Baffy G, Brunt EM and Caldwell SH:
Hepatocellular carcinoma in non-alcoholic fatty liver disease: an
emerging menace. J Hepatol. 56:1384–1391. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bertz J, Zang C, Liu H, Wächter M,
Possinger K, Koeffler HP and Elstner E: Compound 48, a novel dual
PPAR alpha/gamma ligand, inhibits the growth of human CML cell
lines and enhances the anticancer-effects of imatinib. Leuk Res.
33:686–692. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han S, Ritzenthaler JD, Zheng Y and Roman
J: PPARbeta/delta agonist stimulates human lung carcinoma cell
growth through inhibition of PTEN expression: the involvement of
PI3K and NF-kappaB signals. Am J Physiol Lung Cell Mol Physiol.
294:L1238–L1249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Geng J, Li X, Lang X, Qiao C, Hu M, Yang
J, et al: Combination of cetuximab and rapamycin enhances the
therapeutic efficacy in hepatocellular carcinoma. Technol Cancer
Res Treat. 13:377–385. 2014.PubMed/NCBI
|
|
16
|
Yang P, Zhang Y, Pang J, Zhang S, Yu Q, He
L, Wagner KU, Zhou Z and Wang CY: Loss of Jak2 impairs endothelial
function by attenuating Raf-1/MEK1/Sp-1 signaling along with
altered eNOS activities. Am J Pathol. 183:617–625. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mao F, Zhang L, Cai MH, Guo H and Yuan HH:
Leonurine hydrochloride induces apoptosis of H292 lung cancer cell
by a mitochondria-dependent pathway. Pharm Biol. 9:1–7. 2015.(Epub
ahead of print).
|
|
18
|
Yaidikar L and Thakur S: Punicalagin
attenuated cerebral ischemia-reperfusion insult via inhibition of
proinflammatory cytokines, up-regulation of Bcl-2, down-regulation
of Bax, and caspase-3. Mol Cell Biochem. 402:141–148. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu JJ, Hu T, Wu XY, Wang CZ, Xu Y, Zhang
Y, et al: Peroxisome proliferator-activated receptor-gamma agonist
rosiglitazone-induced apoptosis in leukemia k562 cells and its
mechanisms of action. Int J Toxicol. 28:123–131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu LH, Cheng NS, Xiong XZ and Wei DP:
Effect of PPAR-gamma ligand RGZ on inhibiting the cell
proliferation of cholangiocarcinoma. Sichuan Da Xue Xue Bao Yi Xue
Ban. 38:295–297. 2007.(In Chinese). PubMed/NCBI
|
|
21
|
Ciarcia R, Damiano S, Montagnaro S,
Pagnini U, Ruocco A, Caparrotti G, et al: Combined effects of PI3K
and SRC kinase inhibitors with imatinib on intracellular calcium
levels, autophagy and apoptosis in CML-PBL cells. Cell Cycle.
12:2839–2848. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sawayama H, Ishimoto T, Watanabe M,
Yoshida N, Sugihara H, Kurashige J, et al: Small molecule agonists
of PPAR-γ exert therapeutic effects in esophageal cancer. Cancer
Res. 74:575–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kulkarni S, Sitaru C, Jakus Z, Anderson
KE, Damoulakis G, Davidson K, et al: PI3Kβ plays a critical role in
neutrophil activation by immune complexes. Sci Signal.
4:ra232011.PubMed/NCBI
|
|
24
|
Koyasu S: The role of PI3K in immune
cells. Nat Immunol. 4:313–319. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J: Roles of the PI3K/Akt pathway in
Epstein-Barr virus-induced cancers and therapeutic implications.
World J Virol. 1:154–161. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang YJ, Zhang AQ, Zhao XX, Tian ZL and
Yao L: Nicorandil protects against ischaemia-reperfusion injury in
newborn rat kidney. Pharmacology. 92:245–256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fujita M, Yagami T, Fujio M, Tohji C,
Takase K, Yamamoto Y, et al: Cytotoxicity of troglitazone through
PPARγ-independent pathway and p38 MAPK pathway in renal cell
carcinoma. Cancer Lett. 312:219–227. 2011. View Article : Google Scholar : PubMed/NCBI
|