Introduction
Malignant pleural mesothelioma (MPM) is a tumor
derived from the mesothelial cells lining the pleural spaces. MPM
has highly invasive and aggressive clinical characteristics.
Approximately 80% of MPM patients have a history of occupational
asbestos exposure, which is considered to be a risk factor for the
development of the disease (1). The
molecular pathogenesis of MPM is not well understood. The most
common mutations in MPMs are losses in 9p21, 1p36, 14q32 and 22q12,
and gains in 5p, 7p and 8q24, which have been detected by
comparative genomic hybridization analysis (2,3).
Homozygous deletion of the 9p21 locus encoding two critical
cyclin-dependent kinase inhibitors, p16INK4a and
p15INK4b, have been reported in up to 80% of MPMs, and
this mutation may be of diagnostic utility (4,5). The tumor
suppressor neurofibromin 2 is encoded by the NF2 gene,
located on chromosome 22q12. Mutations in NF2 are found in
~40% of MPMs, and heterozygous loss of NF2 is identified in
~74% of MPMs (6,7). Mutations are rare in the TP53 and
RAS genes, which are frequently present in epithelial solid
tumors (8,9). Epigenetic alterations, such as DNA
methylation, have been found in MPMs, which have a different
profile compared with lung cancer (10–12). MPMs,
particularly of the epithelioid subtype, may be hard to
differentiate from adenocarcinoma arising in the lung periphery,
and epidemiological evidence indicates that asbestos and smoking
are shared risk factors for these diseases (2,13,14). Currently, the differential diagnosis
of MM is based on a range of morphological analyses, including a
combination of histological and immunohistochemical staining, and
electron microscopy (13,15,16).
Cytogenetic studies have been performed on MPMs and
adenocarcinomas arising in the lung periphery, however, no
chromosomal aberrations specific to either of the tumor types have
been identified (2,14).
Reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) is a method for evaluating DNA copy number
changes, including losses, gains and amplifications of DNA
sequences (17–19). Copy number gains (CNGs) of EGFR
and KRAS have been observed in lung cancer, particularly in
adenocarcinoma (18,20). Furthermore, CNGs of FGFR1 and
SOX2 have been observed in lung cancer, particularly in
squamous cell carcinoma (21–25). c-Met was recently reported to be
activated in MPM by overexpression or mutations in MET
(26), and MET amplification
is a known cause of resistance to EGFR-tyrosine kinase
inhibitor (TKI) treatment in lung cancer (27). RT-qPCR was used in the present study
on 83 primary MPM and 53 primary lung adenocarcinomas to compare
the CNGs of EGFR, KRAS, MET, FGFR1 and SOX2.
Materials and methods
Tumor samples
Surgically resected specimens of 53 lung
adenocarcinomas and 83 MPMs (57 epithelioid, 8 sarcomatoid, 15
biphasic, 2 desmoplastic and 1 lymphohistiocytic) were obtained.
All the lung adenocarcinomas and 11 of the MPM samples were
obtained from Okayama University Hospital (Okayama, Japan). Another
18 MPMs were obtained from Yamaguchi-Ube Medical Center (Ube,
Japan), 2 were obtained from Okayama Rosai Hospital (Okayama,
Japan) and the remaining 52 were obtained from Karmanos Cancer
Center (Detroit, MI, USA). All Japanese samples were collected
between March 2002 and September 2011, and all samples from the USA
were collected >10 years ago. Resected tumors were stored at
−80°C until DNA extraction. Permission from the Institutional
Review Board and informed consent were obtained at each collection
site.
DNA extraction
Genomic DNA was obtained from primary tumors by
standard phenol:chloroform (1:1) extraction, followed by ethanol
precipitation, or using a DNeasy Tissue kit (Qiagen, Inc.,
Valencia, CA, USA).
RT-qPCR for copy number
evaluation
CNGs of EGFR, KRAS, MET, FGFR1 and
SOX2 genes were determined by RT-qPCR assays using Power
SYBR® Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA,
USA), as previously described (18,19).
Briefly, samples of 1 µl were analyzed per assay using with StepOne
Plus Real-Time PCR System (Themo Fisher Scientific). PCR conditions
were initial denaturation at 95°C for 10 min followed by 40 cycles
of amplification at 95°C for 15 sec and 60°C for 60 sec. The
samples were analyzed in triplicate using StepOne Plus RT PCR
software (version 2.0; Themo Fisher Scientific) and the
LINE1 gene was used as a reference gene for all copy number
analyses, as this is the most abundant autonomous retrotransposon
in the human genome, constituting 17%. Each amplification reaction
was checked for the absence of non-specific PCR products by
performing a melting curve analysis. The copy number calculation
was conducted using the comparative cycle threshold (Ct) method
following validation of the PCR reaction efficiency of EGFR,
KRAS, MET, FGFR1, SOX2 and LINE1. The PCR primer
sequences for EGFR, KRAS, MET and LINE1 primers have
previously been described (17–19). The
PCR primer sequences for FGFR1 and SOX2 were designed
by Primer 3 plus software and by modification of the sequences. The
PCR primer sequences were as follows: FGFR1 forward,
5′-AGCCACCACATGGCATACTT-3′ and reverse, 5′-GGTGACAAGGCTCCACATCT-3′;
and SOX2 forward, 5′-CGTCACATGGATGGTTGTCT-3′ and reverse,
5′-GCCGCCGATGATTGTTATTA-3′. The relative copy number of each sample
was determined by comparing the ratio of the target gene to
LINE1 in each sample with the ratio of these genes in normal
human genomic DNA (EMD Biosciences, Darmstadt, Germany) prepared
from a mixture of human blood cells from 6–8 donors, as a diploid
control. Our previous study defined a copy number of ≥4 as a gene
gain in cell lines (17,18). However, considering the contamination
by non-malignant cells in primary samples (estimated mean per
tumor, 50% tumor cells and 50% non-malignant cells), the cut-off
value of 3 copy numbers rather than 4 was used for primary tumors
in this study (17).
Detection of EGFR mutations
The EGFR mutational status was determined
using a PCR-based length polymorphism and restriction fragment
length polymorphism assay, as previously described (28). Briefly, the common deletions of exon
19 were distinguished from the wild-type based on PCR product
length polymorphisms using 12% polyacrylamide gel electrophoresis
(PAGE) and ethidium bromide staining. For the exon 21 L858R
mutation, Sau96I digestion, which specifically digests the
mutant type, was performed prior to 12% PAGE.
Statistical analyses
Differences between the two groups were assessed
using the χ2 test or Fisher's exact test as required.
All data were analyzed using JMP software version 9.0.0 (SAS
Institute Inc., Cary, NC, USA). For all analyses, P<0.05 was
considered to indicate a statistically significant difference.
Results
CNGs in MPMs and lung
adenocarcinomas
In the 83 MPM samples, the CNGs of EGFR, KRAS,
MET, FGFR1 and SOX2 were detected in 12 (14.5%), 8
(9.6%), 5 (6.0%), 4 (4.8%), and 1 (1.2%) of the samples,
respectively. In the epithelioid subtype of MPM (n=57), the CNGs of
EGFR, KRAS, MET, FGFR1 and SOX2 were detected in 7
(12.3%), 5 (8.8%), 3 (5.3%), 4 (7.0%) and 0 (0.0%) of the samples,
respectively. In the other subtypes of MPMs (n=26), the CNGs of
EGFR, KRAS, MET, FGFR and SOX2 were detected in 5
(19.2%), 3 (11.5%), 2 (7.7%), 0 (0%) and 1 (3.8%) of the samples,
respectively. In the 53 lung adenocarcinomas, the CNGs of EGFR,
KRAS, MET, FGFR1 and SOX2 were detected in 21 (39.6%),
12 (22.6%), 5 (9.4%), 10 (18.9%) and 0 (0.0%) of the samples,
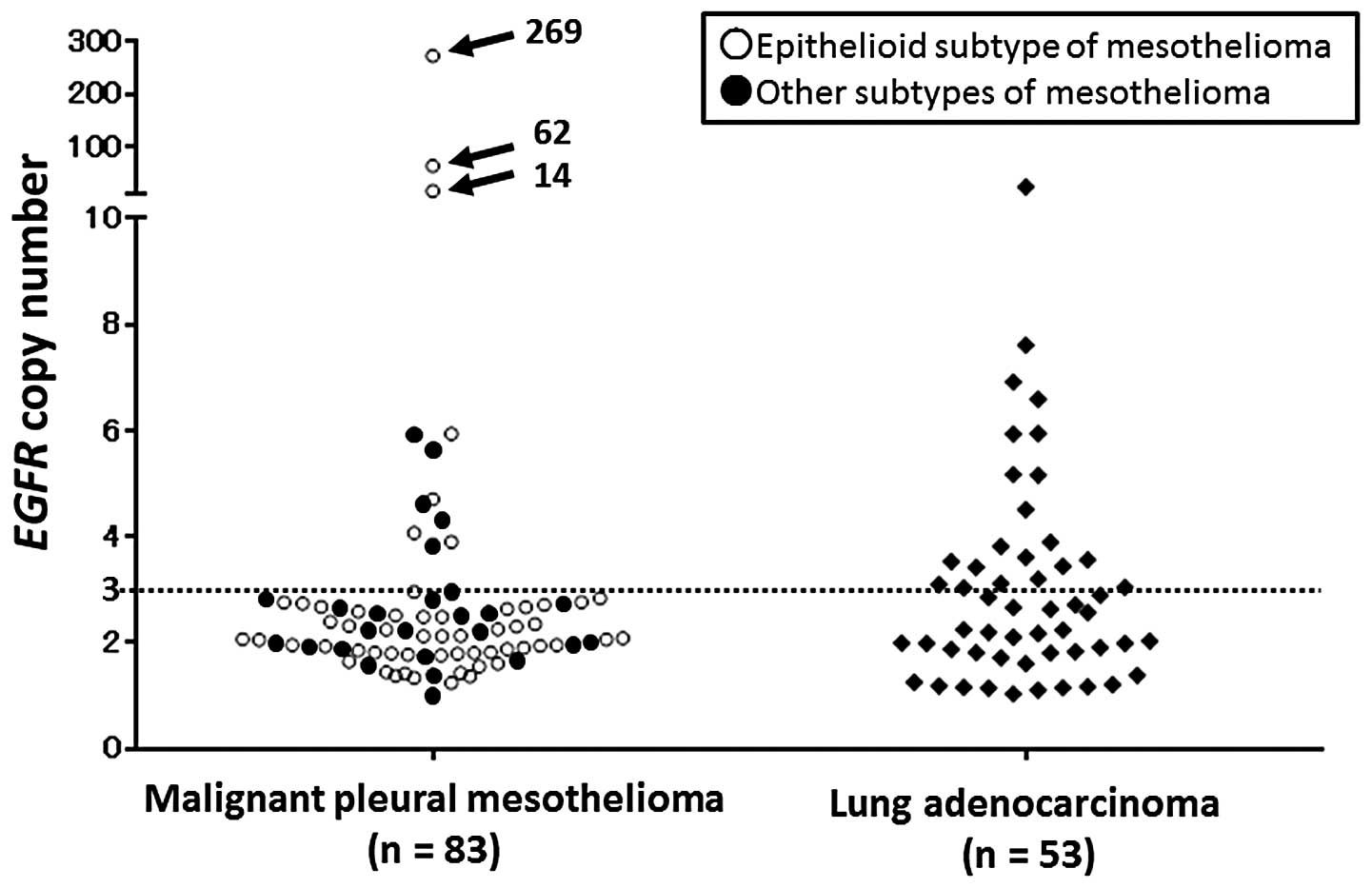
respectively (Table I; Fig. 1). Three cases of MPMs were
demonstrated to have numerous CNGs of EGFR (269, 62 and 14,
respectively). The CNGs of EGFR, KRAS and FGFR1 were
significantly less frequent in the MPMs compared with the lung
adenocarcinomas (P=0.0018, 0.048 and 0.018, respectively). In the
epithelioid subtype of MPMs, the CNGs of EGFR were
significantly less frequent than those in the lung adenocarcinomas
(P=0.0018), and in other subtypes of MPMs, the CNGs of FGFR1
were significantly less frequent compared with those of the lung
adenocarcinomas (P=0.026). In the MPMs, an absence and presence of
CNGs were observed in 64 (77.1%) and 19 (22.9%) of the 83 cases,
respectively. In the epithelioid MPMs, absent/present CNGs were
observed in 47 (82.5%) and 10 (17.5%) of the 57 cases,
respectively. In the other subtypes of the MPMs, the absence and
presence of CNGs were observed in 17 (65.4%) and 9 (34.6%) of the
26 cases, respectively. In the lung adenocarcinomas, the absence
and presence of CNGs were observed in 24 (45.3%) and 29 (54.7%) of
the 53 cases, respectively (Table
II). The MPMs and the epithelioid subtypes of the MPMs had less
frequent CNGs than the lung adenocarcinomas (P=0.0002 and P=0.0001,
respectively).
 | Table I.CNGs of EGFR, KRAS, MET, FGFR1
and SOX2 in MPMs and lung adenocarcinomas. |
Table I.
CNGs of EGFR, KRAS, MET, FGFR1
and SOX2 in MPMs and lung adenocarcinomas.
|
| MPMs (n=83)
|
|
|
|---|
|
|
|
| Epithelioid | Other | Lung |
|
|---|
|
| All (n=83)
| subtype (n=57)
| subtypes (n=26)
| adenocarcinoma
(n=53)
|
|---|
| Genes | No. | % | No. | % | No. | % | No. | % |
|---|
| EGFR | 12a | 14.5 | 7a | 12.3 | 5 | 19.2 | 21 | 39.6 |
| KRAS |
8b |
9.6 | 5 |
8.8 | 3 | 11.5 | 12 | 22.6 |
| MET | 5 |
6.0 | 3 |
5.3 | 2 |
7.7 | 5 |
9.4 |
| FGFR1 |
4b |
4.8 | 4 |
7.0 |
0b |
0.0 | 10 | 18.9 |
| SOX2 | 1 |
1.2 | 0 |
0.0 | 1 |
3.8 | 0 |
0.0 |
| Table II.Frequency of the absence or presence
of CNGs in MPMs and lung adenocarcinomas. |
Table II.
Frequency of the absence or presence
of CNGs in MPMs and lung adenocarcinomas.
|
| Absence of CNGs
| Presence of CNGs
|
|---|
| Cancer type | No. | % | No. | % |
|---|
| Malignant pleural
mesothelioma (n=83)a |
64a | 77.1 | 19 | 22.9 |
| Epithelioid subtype
(n=57)a |
47a | 82.5 | 10 | 17.5 |
| Other subtypes
(n=26) | 17 | 65.4 | 9 | 34.6 |
| Lung adenocarcinoma
(n=53) | 24 | 45.3 | 29 | 54.7 |
EGFR mutations
No EGFR mutation was detected in the 83 MPMs.
In the lung adenocarcinomas, EGFR mutations were detected in
21 (39.6%) cases; 14 cases exhibited an exon 19 deletion and 7
cases exhibited an exon 21 mutation (L858R).
Discussion
The main finding of the present study is that the
pattern of DNA CNGs of MPM is different from that in lung
adenocarcinoma. MPMs exhibited less CNGs of the genes examined in
compared with the lung adenocarcinomas. The epithelioid subtype of
MPM, which is often difficult to distinguish from lung
adenocarcinoma, similarly exhibited these CNGs less frequently
compared with the lung adenocarcinomas. To the best of our
knowledge, only a limited number of studies have previously
analyzed the presence and frequency of EGFR CNGs in MPMs
(2,29–32), and
no studies have focused on CNGs of KRAS, MET, FGFR1 or
SOX2 in MPM. A large number of samples (n=83) were screened
in the present study, whereas the previous studies were based on
smaller sample sizes and may have underestimated the true frequency
of such CNGs.
Although CNGs of SOX2 were seldom observed in
the MPMs and lung adenocarcinomas, the CNGs of the remaining four
genes were detected in the MPM samples to a certain extent. The
fact that the CNGs of four genes in the MPMs were less frequent in
comparison to the lung adenocarcinomas suggested that CNG may not
be a pivotal mechanism for the activation of oncogenes in MPMs, and
that different mechanisms may be of greater importance. It has been
previously reported that EGFR is overexpressed in 60–70% of
MPM tissue specimens; however, it is not overexpressed in the
normal mesothelium (29,33). Furthermore, exposure to asbestos
fibers is known to cause EGFR aggregation (34). In the present study, EGFR,
located at 7p12-p13, was the most frequent gene to exhibit CNGs (12
out of 83 MPMs and 20 out of 53 lung adenocarcinomas). Bjorkqvist
et al (2) reported similar
results, such as gains of genetic material in 5p, 6p and 7p between
MPMs and lung adenocarcinomas. The study detected a gain in 7p in 7
out of 34 MPMs and 11 out of 30 lung adenocarcinomas (2). MPMs rarely harbor EGFR mutations
(31,35–37). There
were no EGFR mutations detected in MPMs in the present
study, as expected. Upon analysis, three cases of MPMs exhibited
high EGFR gene amplification (CNG>10), and these cases
were all epithelioid MPMs, which was consistent with the previous
studies by Okuda et al (29)
and Enomoto et al (31). It
remains unclear whether high-level amplification of EGFR is
more prominent in MPMs compared with lung adenocarcinomas, although
the frequency of CNGs for EGFR is lower in MPMs compared
with lung adenocarcinomas. In MPMs with EGFR amplification,
the inhibition of EGFR pathways should exert an antitumor
effect. In lung cancer, the results of two randomized phase III
trials that compared a placebo to erlotinib or gefitinib treatment
indicated that EGFR copy number detected by fluorescence
in situ hybridization was the best predictor of survival
(38). Patients with colorectal
cancer who responded to anti-EGFR treatment with cetuximab
or panitumumab exhibited an increased EGFR copy number
(39). Although two phase II studies
of single-agent EGFR-TKI therapy to treat MPMs failed to
demonstrate their clinical efficacy, in the gefitinib trial, 2 of
43 MPM patients responded to gefitinib (40,41). These
data suggest that a small proportion of patients (with EGFR
gene amplification) may be candidates for anti-EGFR
treatment (29).
In conclusion, the present study detected novel CNGs
in genes other than EGFR. MPM samples exhibited these CNGs
less frequently compared with lung adenocarcinomas. The differences
in DNA CNG between the two tumor types suggested that they are
genetically different.
Acknowledgements
The authors would like to thank Ms. Fumiko Isobe for
providing excellent technical support. The study received
Grant-in-Aids for the 13 fields of occupational injuries and
illnesses of the Japan Labor Health and Welfare Organization.
References
|
1
|
Spirtas R, Heineman EF, Bernstein L, Beebe
GW, Keehn RJ, Stark A, Harlow BL and Benichou J: Malignant
mesothelioma: Attributable risk of asbestos exposure. Occup Environ
Med. 51:804–811. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Björkqvist AM, Tammilehto L, Nordling S,
Nurminen M, Anttila S, Mattson K and Knuutila S: Comparison of DNA
copy number changes in malignant mesothelioma, adenocarcinoma and
large-cell anaplastic carcinoma of the lung. Br J Cancer.
77:260–269. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taniguchi T, Karnan S, Fukui T, Yokoyama
T, Tagawa H, Yokoi K, Ueda Y, Mitsudomi T, Horio Y, Hida T, et al:
Genomic profiling of malignant pleural mesothelioma with
array-based comparative genomic hybridization shows frequent
non-random chromosomal alteration regions including JUN
amplification on 1p32. Cancer Sci. 98:438–446. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chiosea S, Krasinskas A, Cagle PT,
Mitchell KA, Zander DS and Dacic S: Diagnostic importance of 9p21
homozygous deletion in malignant mesotheliomas. Mod Pathol.
21:742–747. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Toyooka S, Kishimoto T and Date H:
Advances in the molecular biology of malignant mesothelioma. Acta
Med Okayama. 62:1–7. 2008.PubMed/NCBI
|
|
6
|
Fleury-Feith J, Lecomte C, Renier A,
Matrat M, Kheuang L, Abramowski V, Levy F, Janin A, Giovannini M
and Jaurand MC: Hemizygosity of Nf2 is associated with increased
susceptibility to asbestos-induced peritoneal tumours. Oncogene.
22:3799–3805. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nemoto H, Tate G, Kishimoto K, Saito M,
Shirahata A, Umemoto T, Matsubara T, Goto T, Mizukami H, Kigawa G,
et al: Heterozygous loss of NF2 is an early molecular alteration in
well-differentiated papillary mesothelioma of the peritoneum.
Cancer Genet. 205:594–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Metcalf RA, Welsh JA, Bennett WP, et al:
p53 and Kirsten-ras mutations in human mesothelioma cell lines.
Cancer Res. 52:2610–2615. 1992.PubMed/NCBI
|
|
9
|
Papp T, Schipper H, Pemsel H, et al:
Mutational analysis of N-ras, p53, p16INK4a, p14ARF and CDK4 genes
in primary human malignant mesotheliomas. Int J Oncol. 18:425–433.
2001.PubMed/NCBI
|
|
10
|
Toyooka S, Pass HI, Shivapurkar N,
Fukuyama Y, et al: Aberrant methylation and simian virus 40 tag
sequences in malignant mesothelioma. Cancer Res. 61:5727–5730.
2001.PubMed/NCBI
|
|
11
|
Kobayashi N, Toyooka S, Yanai H, et al:
Frequent p16 inactivation by homozygous deletion or methylation is
associated with a poor prognosis in Japanese patients with pleural
mesothelioma. Lung Cancer. 62:120–125. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goto Y, Shinjo K, Kondo Y, et al:
Epigenetic profiles distinguish malignant pleural mesothelioma from
lung adenocarcinoma. Cancer Res. 69:9073–9082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Addis B and Roche H: Problems in
mesothelioma diagnosis. Histopathology. 54:55–68. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gee GV, Koestler DC, Christensen BC, et
al: Downregulated microRNAs in the differential diagnosis of
malignant pleural mesothelioma. Int J Cancer. 127:2859–2869. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Betta PG, Magnani C, Bensi T, Trincheri NF
and Orecchia S: Immunohistochemistry and molecular diagnostics of
pleural malignant mesothelioma. Arch Pathol Lab Med. 136:253–261.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Husain AN, Colby T, Ordonez N, Krausz T,
Attanoos R, Beasley MB, Borczuk AC, Butnor K, Cagle PT, Chirieac
LR, et al: International Mesothelioma Interest Group: Guidelines
for pathologic diagnosis of malignant mesothelioma: 2012 update of
the consensus statement from the International Mesothelioma
Interest Group. Arch Pathol Lab Med. 137:647–667. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamamoto H, Shigematsu H, Nomura M,
Lockwood WW, Sato M, Okumura N, Soh J, Suzuki M, Wistuba II, Fong
KM, et al: PIK3CA mutations and copy number gains in human lung
cancers. Cancer Res. 68:6913–6921. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soh J, Okumura N, Lockwood WW, Yamamoto H,
Shigematsu H, Zhang W, Chari R, Shames DS, Tang X, MacAulay C, et
al: Oncogene mutations, copy number gains and mutant allele
specific imbalance (MASI) frequently occur together in tumor cells.
PLoS One. 4:e74642009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kubo T, Yamamoto H, Lockwood WW, Valencia
I, Soh J, Peyton M, Jida M, Otani H, Fujii T, Ouchida M, et al:
MET gene amplification or EGFR mutation activate
MET in lung cancers untreated with EGFR tyrosine
kinase inhibitors. Int J Cancer. 124:1778–1784. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sasaki H, Hikosaka Y, Kawano O, Moriyama
S, Yano M and Fujii Y: Evaluation of Kras gene mutation and copy
number gain in non-small cell lung cancer. J Thorac Oncol. 6:15–20.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao X, Weir BA, LaFramboise T, Lin M,
Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG,
et al: Homozygous deletions and chromosome amplifications in human
lung carcinomas revealed by single-nucleotide polymorphism array
analysis. Cancer Res. 65:5561–5570. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan P, Kadara H, Behrens C, Tang X, Woods
D, Solis LM, Huang J, Spinola M, Dong W, Yin G, et al:
Sex-determining region Y-Box 2 (SOX2) is a potential
cell-lineage gene highly expressed in the pathogenesis of squamous
cell carcinomas of the lung. PLoS One. 5:e91122010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kohler LH, Mireskandari M, Knösel T,
Altendorf-Hofmann A, Kunze A, Schmidt A, Presselt N, Chen Y and
Petersen I: FGFR1 expression and gene copy numbers in human
lung cancer. Virchows Arch. 461:49–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Heist RS, Mino-Kenudson M, Sequist LV,
Tammireddy S, Morrissey L, Christiani DC, Engelman JA and Iafrate
AJ: FGFR1 amplification in squamous cell carcinoma of the
lung. J Thorac Oncol. 7:1775–1780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sasaki H, Yokota K, Hikosaka Y, Moriyama
S, Yano M and Fujii Y: Increased SOX2 copy number in lung
squamous cell carcinomas. Exp Ther Med. 3:44–48. 2012.PubMed/NCBI
|
|
26
|
Jagadeeswaran R, Ma PC, Seiwert TY,
Jagadeeswaran S, Zumba O, Nallasura V, Ahmed S, Filiberti R,
Paganuzzi M, Puntoni R, et al: Functional analysis of c-met
hepatocyte growth factor pathway in malignant pleural mesothelioma.
Cancer Res. 66:352–361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in
lung cancer by activating ERBB3 signaling. Science. 316:1039–1043.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Asano H, Toyooka S, Tokumo M, Ichimura K,
Aoe K, Ito S, Tsukuda K, Ouchida M, Aoe M, Katayama H, et al:
Detection of EGFR gene mutation in lung cancer by
mutant-enriched polymerase chain reaction assay. Clin Cancer Res.
12:43–48. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Okuda K, Sasaki H, Kawano O, Yukiue H,
Yokoyama T, Yano M and Fujii Y: Epidermal growth factor receptor
gene mutation, amplification and protein expression in malignant
pleural mesothelioma. J Cancer Res Clin Oncol. 134:1105–1111. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rena O, Boldorini LR, Gaudino E and
Casadio C: Epidermal growth factor receptor overexpression in
malignant pleural mesothelioma: Prognostic correlations. J Surg
Oncol. 104:701–705. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Enomoto Y, Kasai T, Takeda M, Takano M,
Morita K, Kadota E, Iizuka N, Maruyama H, Haratake J, Kojima Y, et
al: A comparison of epidermal growth factor receptor expression in
malignant peritoneal and pleural mesothelioma. Pathol Int.
62:226–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Takeda M, Kasai T, Enomoto Y, Takano M,
Morita K, Kadota E, Iizuka N, Maruyama H and Nonomura A: Genomic
gains and losses in malignant mesothelioma demonstrated by FISH
analysis of paraffin-embedded tissues. J Clin Pathol. 65:77–82.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Destro A, Ceresoli GL, Falleni M, Zucali
PA, Morenghi E, Bianchi P, Pellegrini C, Cordani N, Vaira V,
Alloisio M, et al: EGFR overexpression in malignant pleural
mesothelioma. An immunohistochemical and molecular study with
clinico-pathological correlations. Lung Cancer. 51:207–215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pache JC, Janssen YM, Walsh ES, Quinlan
TR, Zanella CL, Low RB, Taatjes DJ and Mossman BT: Increased
epidermal growth factor-receptor protein in a human mesothelial
cell line in response to long asbestos fibers. Am J Pathol.
152:333–340. 1998.PubMed/NCBI
|
|
35
|
Cortese JF, Gowda AL, Wali A, Eliason JF,
Pass HI and Everson RB: Common EGFR mutations conferring
sensitivity to gefitinib in lung adenocarcinoma are not prevalent
in human malignant mesothelioma. Int J Cancer. 118:521–522. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Velcheti V, Kasai Y, Viswanathan AK,
Ritter J and Govindan R: Absence of mutations in the epidermal
growth factor receptor (EGFR) kinase domain in patients with
mesothelioma. J Thorac Oncol. 4:5592009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mezzapelle R, Miglio U, Rena O, Paganotti
A, Allegrini S, Antona J, Molinari F, Frattini M, Monga G, Alabiso
O, et al: Mutation analysis of the EGFR gene and downstream
signalling pathway in histologic samples of malignant pleural
mesothelioma. Br J Cancer. 108:1743–1749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tsao MS, Sakurada A, Cutz JC, Zhu CQ,
Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, et
al: Erlotinib in lung cancer - molecular and clinical predictors of
outcome. N Engl J Med. 353:133–144. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Moroni M, Veronese S, Benvenuti S,
Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M,
Siena S and Bardelli A: Gene copy number for epidermal growth
factor receptor (EGFR) and clinical response to
antiEGFR treatment in colorectal cancer: a cohort study.
Lancet Oncol. 6:279–286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Govindan R, Kratzke RA, Herndon JE,
Niehans GA, Vollmer R, Watson D, Green MR and Kindler HL: Cancer
and Leukemia Group B (CALGB 30101): Gefitinib in patients with
malignant mesothelioma: A phase II study by the Cancer and Leukemia
Group B. Clin Cancer Res. 11:2300–2304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Garland LL, Rankin C, Gandara DR, Rivkin
SE, Scott KM, Nagle RB, Klein-Szanto AJ, Testa JR, Altomare DA and
Borden EC: Phase II study of erlotinib in patients with malignant
pleural mesothelioma: A Southwest Oncology Group Study. J Clin
Oncol. 25:2406–2413. 2007. View Article : Google Scholar : PubMed/NCBI
|