Introduction
Chronic myeloid leukemia (CML) is a hematopoietic
stem cell disorder, characterized by the expression and abnormal
tyrosine kinase activity of the Bcr-Abl protein (1). The current recommended first line
therapy for patients with newly diagnosed CML, is imatinib.
However, resistance to imatinib is a significant problem, despite
the good clinical results achieved with this tyrosine kinase
inhibitor in the treatment of CML (2). Therefore, the identification of novel
therapeutic agents, in addition to an understanding of their
underlying mechanisms, is necessary in order to improve the outcome
in patients with CML.
Histone deacetylase inhibitors (HDACi) represent a
novel class of anticancer agents that are currently under
investigation in preclinical models and in phase I–III clinical
trials (3,4). Suberoylanilide hydroxamic acid (SAHA) is
an orally bioavailable, well-tolerated pan-HDACi, which exhibits
strong anticancer activity in hematological and solid malignancies
(3–7).
Recently, SAHA has been shown to exhibit anti-CML activity, when
administered either alone or in combination with other agents
(8–10).
Proteasome inhibitors represent a relatively
recently identified class of antineoplastic agents, which function
by interfering with the catalytic 20S core of the proteasome,
thereby preventing the elimination of diverse cellular proteins
that have been targeted for degradation. There are a number of
classes of proteasome inhibitors, including peptide aldehydes, such
as MG-132 (11,12). Recently, it has been reported that
certain proteasome inhibitors, such as bortezomib (also termed
velcade, MG-341 and PS-341), inhibit survival and induce apoptosis
in imatinib-resistant Bcr-Abl cells (13). Furthermore, enhanced lethality towards
CML cells, of a regimen combining HDACi and proteasome inhibitors,
has also been described (11).
Evidence that HDACi promote the antileukemic
activity of certain proteasome inhibitors, particularly in CML
cells (11,14), led to the hypothesis that SAHA and
MG-132 may function synergistically to kill CML cells. Previous
studies have indicated that the combination of SAHA and MG-132 may
induce cell death in imatinib-sensitive K562 cells (11). However, the mechanism underlying this
effect remains unknown and, to the best of our knowledge, there is
no data on combined SAHA and MG-132 treatment in imatinib-resistant
CML cells. The present study investigated the combined effects of
SAHA and MG-132, in imatinib-sensitive K562 cells and
imatinib-resistant K562G cells in vitro, and attempted to
elucidate the mechanisms underlying these effects.
Materials and methods
Cell culture and reagents
The K562 imatinib-sensitive human CML cell line and
the K562G imatinib-resistant human CML cell line were maintained in
RPMI 1640 medium (HyClone Laboratories, Inc., Logan, UT, USA) with
10% fetal bovine serum (HyClone Laboratories, Inc.) at 37°C in a 5%
CO2 humid atmosphere, as previously described (15). SAHA was obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). MG-132 and the
pan-caspase inhibitor, z-VAD-fmk, were obtained from Sigma-Aldrich
(St. Louis, MO, USA). N-acetyl-L-cysteine (NAC) was obtained from
Beyotime Company (Shanghai, China).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cell viability was determined using an MTT assay. In
brief, K562 or K562G cells were cultured in 96-well plates at a
density of 2×104 cells/ml, then treated with or without
SAHA (0.5–8.0 µM; 24–48 h), MG-132 (200–800 nM; 24–48 h) and
z-VAD-fmk (20 µM; 1 h). Subsequently, 20 µl MTT (Sigma-Aldrich)
solution (5 mg/ml) was added into each well and the cells were
incubated for a further 4 h. The supernatant was removed, 200 µl
dimethyl sulfoxide (Amresco, Solon, OH, USA) was added and the
absorbance at 570 nm was detected using an enzyme-linked
immunosorbent assay plate reader (Model 550; Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Colony formation assay
For the leukemic cell colony formation assays, K562
cells were suspended in Iscove modified Dulbecco's medium (Stem
Cell Technologies, Vancouver, BC, Canada), containing 10% fetal
bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100
µg/ml; Sigma-Aldrich), and were then treated with or without SAHA
(2 µM) and/or MG-132 (200 nM). Subsequently, the cells were plated
in 20% FBS and 0.8% methylcellulose. The cells were cultured 7 days
and observed under a light microscope (Olympus, Tokyo, Japan).
Analysis of ROS generation
Intracellular ROS generation was determined using
the Reactive Oxygen Species Assay kit (Beyotime, Shanghai, China),
according to the manufacturer's instructions. Rosup (50 mg/ml;
Beyotime Institute of Biotechnology, Shanghai, China) was used as a
positive control. Cells were pretreated with or without NAC (2 mM)
for 1 h, followed by co-treatment with SAHA and MG-132 for 24 h.
Cells were then incubated with DCFH-DA (10 µM) for 30 min at 37°C,
washed twice with phosphate-buffered saline (PBS), harvested and
then suspended in PBS. A drop of stained cell suspension was placed
on a slide for the immediate observation of fluorescence, using a
fluorescence microscope (IX71; Olympus Corporation, Tokyo,
Japan).
Western blot analysis
Following administration of SAHA (1–4 µM) for 3–24 h
and MG-132 (200–400 nM) for 6–24 h with or without pretreatment
with NAC (2 mM) for 1 h, cells were washed twice with PBS and lysed
in a radioimmunoprecipitation assay buffer (0.5 M Tris-HCl, pH 7.4;
1.5 M NaCl; 2.5% deoxycholic acid; 10% NP-40; and 10 mM EDTA).
Equal quantities of total cell lysates were separated on sodium
dodecyl-sulfate-polyacrylamide gels containing 6–12% acrylamide
gradients, and then transferred to polyvinylidene difluoride
membranes (Millipore, Bedford, MA, USA). The membranes were blocked
with 5% non-fat milk and incubated with primary antibodies at 4°C
overnight in Tris-buffered saline with Tween-20 (10 mM Tris-HCl, pH
8, 150 mM NaCl, 0.1% Tween-20). The membranes were then incubated
for 2 h with horseradish peroxidase-conjugated goat anti-rabbit
(cat no. 7074) and horse anti-mouse (cat no. 7076) secondary
antibodies purchased from Cell Signaling Technology, Inc. (Beverly,
CA, USA) at a dilution of 1:2,000. The following primary antibodies
were used at a dilution of 1:1,000: Rabbit anti-human polyclonal
c-Abl (cat no. 2862), rabbit anti-human polyclonal
phosphorylated-Bcr (p-Bcr; cat no. 3901), mouse anti-human
monoclonal caspase-9 (cat no. 9508), rabbit anti-human polyclonal
caspase-3 (cat no. 9662), rabbit anti-human monoclonal poly
adenosine diphosphate (ADP)-ribose polymerase (PARP; cat no. 9532)
and rabbit anti-human monoclonal acetylated histone H3 (cat no.
9649), which were obtained from Cell Signaling Technology, Inc.
Anti-c-Abl p-Bcr are subsequently referred to as Bcr-Abl and
p-Bcr-Abl, respectively. Rabbit anti-human polyclonal anti-β-actin
(cat no. sc-7210) was obtained from Santa Cruz Biotechnology, Inc.
and used at a dilution of 1:200. Signals were detected using an ECL
kit (Amersham, Little Chalfont, UK).
Statistical analysis
Data are presented as the mean ± standard deviation.
Data were compared using Student's t-test. All data were analyzed
using SPSS software (version 11.0; SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
SAHA induces cell death, downregulates
expression of Bcr-Abl and upregulates acetylation of histone H3 in
K562 and K562G cells
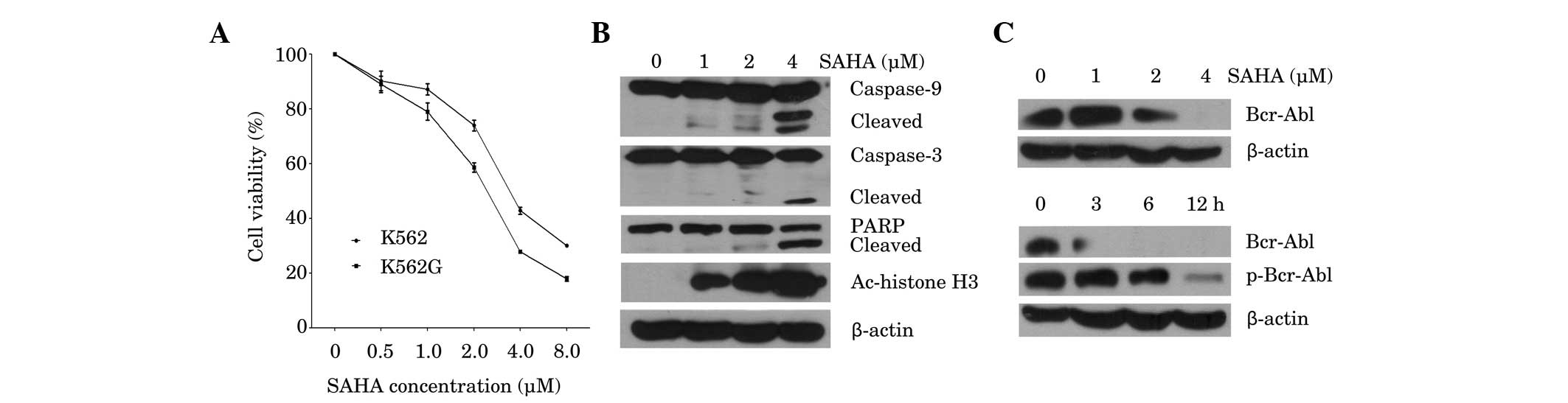
In order to assess the effects of SAHA on cell
viability in imatinib-sensitive K562 cells and imatinib-resistant
K562G cells, cell proliferation was assessed using an MTT assay.
Cells were treated for 48 h with increasing doses of SAHA, from
0.5–8 µM. SAHA inhibited the viability of K562 and K562G cells in a
dose-dependent manner, with 50% inhibition (IC50) values
at 48 h, of 3.93 and 2.42 µM, respectively (Fig. 1A), which indicates that K562 and K562G
cells are both sensitive to SAHA. Subsequently the induction of the
apoptotic pathway by SAHA was investigated. As shown in Fig. 1B, SAHA triggered dose-dependent
cleavage of caspase-9 and caspase-3, followed by PARP cleavage.
Marked induction of apoptosis was observed at the highest dose of
SAHA (4 µM), as evidenced by increased cleavage of caspase-3 and
PARP. In accordance with this result, 4 µM SAHA significantly
inhibited the expression of Bcr-Abl (Fig.
1C; upper panel). Measurement of the protein expression and
phosphorylation status of Bcr-Abl in K562 cells at different time
points following treatment with 4 µM SAHA, demonstrated that
inhibition of Bcr-Abl occurred after 3 h of treatment, while
downregulation of p-Bcr-Abl was observed at 6 h (Fig. 1C; lower panel). Furthermore, exposure
to SAHA for 24 h increased the acetylation of histone H3 in K562
cells in a dose-dependent manner (Fig.
1B). These results indicated that SAHA is cytotoxic to CML
cells, and is associated with downregulation of the Bcr-Abl
oncoprotein and upregulation of histone H3 acetylation.
MG-132 synergistically interacts with
SAHA to induce cell death, inhibit Bcr-Abl and mediate acetylation
of histone H3 in K562 and K562G cells
Subsequently, the combined effects of SAHA and
MG-132 on K562 and K562G cells were examined, using different
combinations of doses of each of these agents (Fig. 2A). Exposure of K562 cells to low dose
SAHA (1 µM) in addition to MG-132 (400 or 800 nM) for 24 h,
resulted in a significant reduction in cell viability compared with
SAHA treatment alone. Similar results were obtained in K562G cells
(Fig. 2A). Further experiments were
conducted in order to determine whether co-treatment with SAHA and
MG-132 altered CML cell colony formation in vitro. The
results demonstrated that treatment of K562 cells with SAHA (2 µM)
or MG-132 (200 nM) alone, inhibited colony formation, compared with
untreated control. However, a combination of SAHA (2 µM) and MG-132
(200 nM), resulted in a greater reduction in colony formation than
that observed with either treatment alone (Fig. 2B). The effects of co-treatment of
cells with SAHA and MG-132 were then examined in association with
the expression of Bcr-Abl, histone acetylation protein and the
apoptotic proteins, caspase-3 and PARP. Western blot analysis was
performed 24 h after treatment with SAHA and MG-132 alone or in
combination. The results demonstrated that treatment with SAHA (1
or 2 µM) or MG-132 (400 nM) alone exerted a minimal effect or no
effect on Bcr-Abl expression. However, K562 cells exposed to
combined treatment, exhibited a marked reduction in Bcr-Abl protein
expression (Fig. 2C). Furthermore,
combined treatment with SAHA (1 or 2 µM) and MG-132 (400 nM)
resulted in a marked increase in the expression of the acetylated
histone H3 protein, compared with either treatment alone. In
accordance with these results, combined treatment resulted in
increased cleavage of caspase-3 and PARP, compared with either
treatment alone (Fig. 2C). The
results in K562G cells also demonstrated that following exposure to
SAHA (1 or 2 µM) and/or MG-132 (200 nM) for 24 h, combined
treatment resulted in a pronounced downregulation of Bcr-Abl
expression, upregulation of acetylated histone H3 expression, and
increase in cleavage of caspase-3 and PARP, compared with either
treatment alone (Fig. 2D).
Furthermore, the pan-caspase inhibitor, z-VAD-fmk, partially
reversed the cell death induced by combined treatment with SAHA and
MG-132, in K562 and K562G cells (Fig.
2E). This suggested an involvement of caspase-independent cell
death induced in combined treatment with SAHA and MG-132.
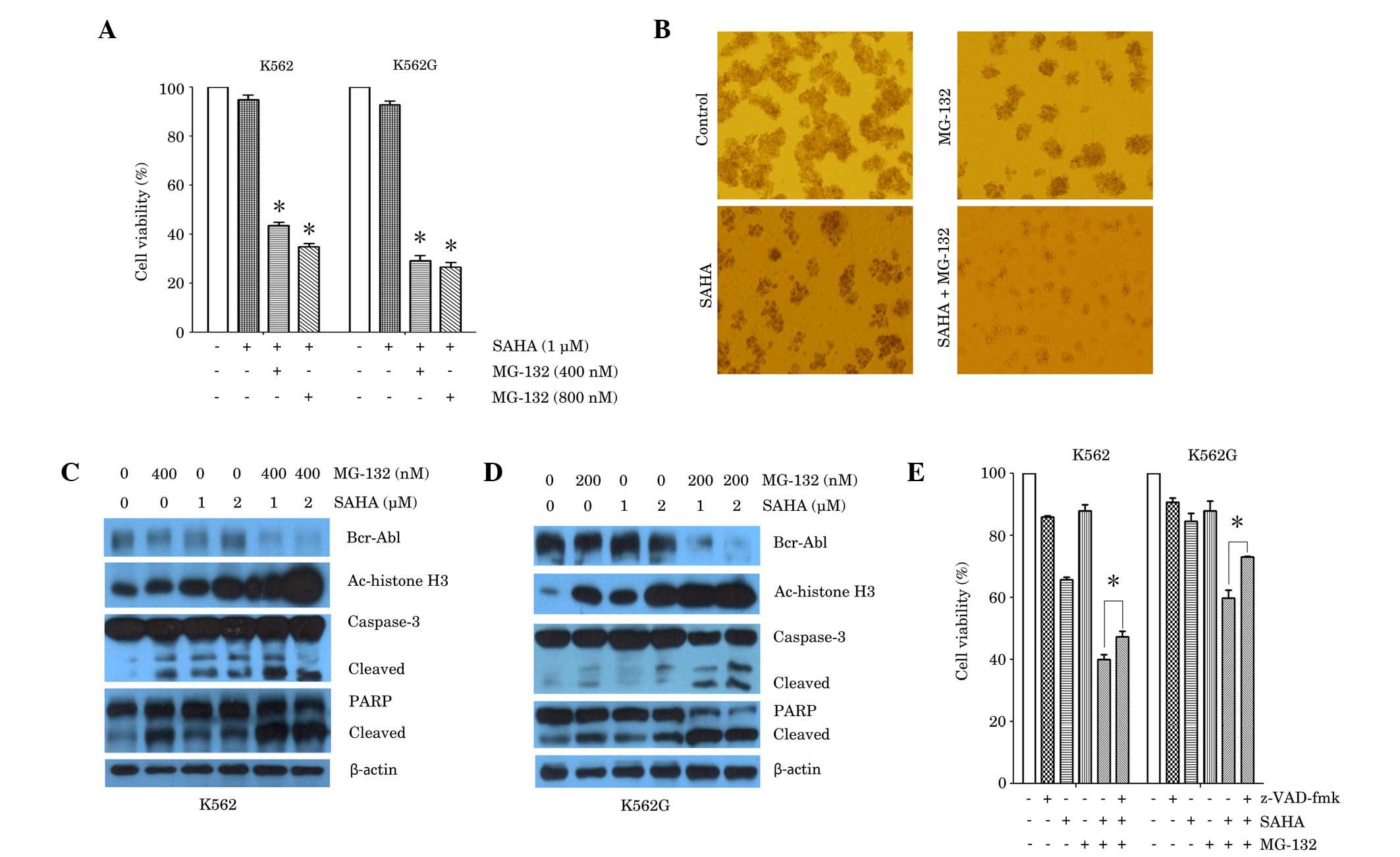 | Figure 2.Synergistic effect of SAHA and MG-132
in the K562 and K562G CML cell lines. (A) K562 cells and K562G
cells were treated with SAHA (1 µM) or SAHA plus MG-132 (400 nM or
800 nM) for 24 h. Cell viability was measured using an MTT assay.
*P<0.0001 vs. SAHA alone. (B) K562 cells were treated with SAHA
(2 µM), MG-132 (200 nM) or SAHA plus MG-132 for 7 days. Colony
formation was observed using a light microscope. Representative
images are shown. (C) K562 cells were treated with SAHA (1 µM or 2
µM), MG-132 (400 nM) or SAHA plus MG-132 (400 nM) for 24 h. Western
blot analysis was performed, using antibodies against Bcr-Abl,
Ac-histone H3, caspase-3 and PARP. β-actin was used as a loading
control. (D) K562G cells were treated with SAHA (1 µM or 2 µM),
MG-132 (200 nM) or SAHA plus MG-132 (200 nM) for 24 h. Western blot
analysis was performed, using antibodies against Bcr-Abl,
Ac-histone H3, caspase-3 and PARP. β-actin was used as a loading
control. Representative blots from three independent experiments
are shown. (E) K562 cells and K562G cells were treated with SAHA (1
µM), MG-132 (200 nM) alone, or SAHA plus MG-132 for 24 h, prior to
which cells were cultured in the presence or absence of z-VAD-fmk
(20 µM) for 1 h. Cell viability was measured using an MTT assay.
*P<0.05. Data are presented as the mean ± standard deviation of
three independent experiments. SAHA, suberoylanilide hydroxamic
acid; CML, chronic myeloid leukemia; Ac-histone, acetylated
histone; PARP, poly adenosine diphosphate-ribose polymerase. |
ROS scavenger NAC reverses the effect
of combined SAHA and MG-132 treatment on cell death, generation of
ROS and Bcr-Abl downregulation, in K562 and K562G cells
It has been reported that SAHA and proteasome
inhibitors, such as MG-132 and NPI-0052, induce ROS-dependent cell
death in cancer cells (14,16). In order to elucidate whether the
generation of ROS by co-treatment with SAHA and MG-132 contributes
to cell death in CML cells, K562 cells were treated with SAHA (1
µM) plus MG-132 (200 nM), with or without pretreatment with the ROS
scavenger, NAC (2 mM). Measurements of intracellular ROS were
determined by fluorescence microscopy, following staining with a
fluorescence probe. Compared with the negative control, combined
treatment with SAHA and MG-132 resulted in a marked increase in the
level of ROS, and this effect was attenuated in the presence of NAC
(Fig. 3A). In accordance with these
results, the MTT assay demonstrated that NAC (2 mM) almost fully
reversed the reduction in cell viability that was induced by
combined treatment with SAHA (1 µM) and MG-132 (200 nM) in K562 and
K562G cells (Fig. 3B). Furthermore,
the reduction in Bcr-Abl expression and in the activation of PARP
induced by SAHA and MG-132, were also reversed by treatment with
NAC (2 mM; Fig. 3C).
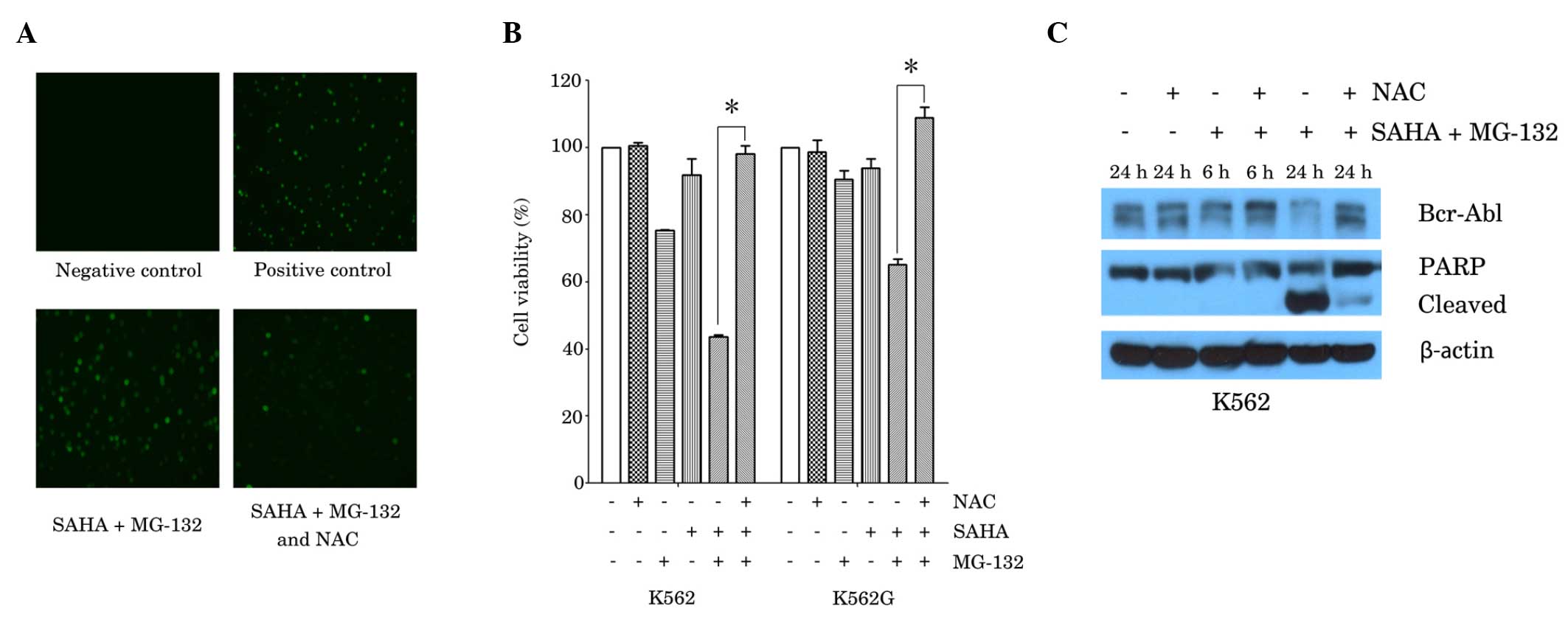 | Figure 3.ROS scavenger, NAC, reversed the
effects of the combination of SAHA and MG-132 in the K562 and K562G
CML cell lines. (A) K562 cells were treated with SAHA (1 µM) plus
MG-132 (200 nM) for 24 h, with or without 1 h pretreatment with NAC
(2 mM), Rosup (50 mg/ml) was used as a positive control. ROS
generation was examined using a fluorescence microscope.
Representative images from three independent experiments are shown.
(B) K562 cells and K562G cells were treated with SAHA (1 µM) or
MG-132 (200 nM) alone, or with SAHA plus MG-132 for 48 h, prior to
which cells were cultured in the presence or absence of NAC (2 mM)
for 1 h. Cell viability was measured using MTT assay. Data are
presented as the mean ± standard deviation of three independent
experiments. *P<0.0001. (C) K562 cells were cultured with SAHA
(1 µM) plus MG-132 (200 nM) for 6 h or 24 h, with or without
pretreatment with NAC (2 mM) for 1 h. Western blot analysis was
performed, using antibodies against Bcr-Abl and PARP. β-actin was
used as a loading control. A representative blot from three
independent experiments is shown. SAHA, suberoylanilide hydroxamic
acid; CML, chronic myeloid leukemia; Ac-histone, acetylated
histone; PARP, poly adenosine diphosphate-ribose polymerase; NAC,
N-acetyl-L-cysteine. |
Discussion
While the introduction of the Bcr-Abl kinase
inhibitor, imatinib, into the clinical armamentarium, represents an
important advance in the treatment of CML, the development of drug
resistance constitutes a significant barrier to curing this disease
(2,17). The present study demonstrated that
treatment with the HDACi, SAHA (0.5–8 µM), induced cell death in
imatinib-sensitive K562 cells, as well as imatinib-resistant K562G
cells, in a dose-dependent manner. The proteasome inhibitor,
MG-132, significantly and synergistically enhanced low-dose
SAHA-induced cell death and inhibited colony formation, suggesting
a synergistic effect of combined SAHA and MG-132 treatment in CML
cells in vitro.
The level of acetylated histones is a useful
intermediary marker of HDACi activity (18). SAHA induces the accumulation of
acetylated histones in the chromatin, and it has been reported that
exposure to SAHA increased the acetylation of histone H3 in K562
cells (8). In accordance with this,
the present study showed that SAHA (1–4 µM) increased the
acetylation of histone H3 in a dose-dependent manner in K562 cells.
Furthermore, combined SAHA and MG-132 treatment resulted in a
significant increase in acetylated histone H3 expression, compared
with SAHA alone, suggesting that MG-132 enhances the activity of
SAHA in CML cells by enhancing histone acetylation.
Bcr-Abl is frequently the causative agent in CML and
is known to be associated with drug resistance (1). It has been reported that SAHA enhances
sensitivity to imatinib in CML cells by decreasing the level of the
Bcr-Abl protein and that this effect is associated with apoptosis
(8). The combination of SAHA with the
proteasome inhibitor, bortezomib, also exhibits an association with
Bcr-Abl downregulation and apoptosis (11). The current study showed that SAHA (2–4
µM) significantly inhibited the expression of Bcr-Abl in CML cells.
This is in accordance with a previous study, which reported that
exposure to 2.0 µM SAHA for 48 h downregulated the expression of
Bcr-Abl in K562 cells, in association with the attenuation of
auto-tyrosine phosphorylation of Bcr-Abl (8). The present study also demonstrated that
co-treatment of SAHA and MG-132 induced significant downregulation
of Bcr-Abl, compared with either drug alone, in K562 and K562G
cells, suggesting that the synergistic effect of SAHA and MG-132 on
cell death in CML cells is associated with Bcr-Abl downregulation.
The mechanisms underlying the development of clinical resistance to
the imatinib, appear to involve either increased expression of the
Bcr-Abl protein through gene amplification (19) or the development of mutations in the
Bcr-Abl catalytic domain, which interfere with imatinib binding to
Bcr-Abl (20). The present finding
that combined treatment with SAHA and MG-132 effectively induces
cell death in the imatinib-resistant K562G cell line, in
association with downregulation of Bcr-Abl expression, raises the
possibility that this combination may be of value in patients
exhibiting the former type of drug resistance. Furthermore, SAHA
(1–4 µM) was shown to activate caspase-9, caspase-3 and PARP
cleavage in K562 cells, in a dose dependent manner. Activation of
caspase-9 and caspase-3 is hypothesized to irreversibly commit a
cell to apoptosis. PARP is the substrate of caspase-3, and
activation of caspase-3 thus commits a cell to apoptosis (21). The present results suggest that
SAHA-induced cell death in CML cells is result of an increase in
apoptosis. Furthermore, MG-132 enhances SAHA-induced caspase
activation in K562 and K562G cells, suggesting that the CML cell
death induced by SAHA in combination with MG-132, is associated
with caspase-dependent apoptosis. However, the pan-caspase
inhibitor, z-VAD-fmk, only resulted in the partial reversal of cell
death of K562 and K562G cells, indicating that caspase-independent
mechanisms also lead to cell death in combined SAHA and MG-132
treatment.
ROS are involved in cell death. They provoke
apoptosis and necrotic cell death (22), and also regulate autophagy (23). The current study demonstrated that the
cell death induced by the combination of SAHA and MG-132, involved
the generation of ROS, and the ROS scavenger, NAC, significantly
blocked cell death, in addition to ROS formation. Activation of the
cleaved form of PARP was also significantly reduced by treatment
with NAC, suggesting that ROS is involved in the induction of cell
death by SAHA and MG-132, and that apoptosis is at least partially
a result of ROS-induced cell death under these conditions. However,
further investigation is required in order to clarify whether other
forms of cell death, such as autophagy and necrosis, lead to cell
the death of CML cells as a result of combined treatment with SAHA
and MG-132.
To date, the association between Bcr-Abl and ROS
remains unclear. A number of reports have suggested that the
increases in ROS generation may stem from Bcr-Abl kinase inhibition
and that ROS levels are increased downstream of Bcr-Abl (24,25).
However, in the present study, NAC reversed the cell death and
Bcr-Abl downregulation induced by combined treatment of SAHA and
MG-132, which indicates that the increase in ROS is upstream of
Bcr-Abl downregulation and mediates the synergistic effect of SAHA
and MG-132 in CML cells.
In conclusion, the present study demonstrates potent
anti-CML activity of combined treatment with SAHA and MG-132 in
vitro and elucidates the underlying mechanisms of this effect,
which are associated with Bcr-Abl downregulation and increased ROS
production. The current study also established ROS as an upstream
regulator of Bcr-Abl. These findings provide a rationale for
further investigation into the use of combined SAHA and MG-132
treatment in CML.
Acknowledgements
This study was supported by the Doctoral Fund of
Ministry of Education of China (grant no. 20120101110010), the
National Natural Science Foundation of China (grant nos. 81070419
and 81200384), Zhejiang Provincial Natural Science Foundation of
China (grant no. R2090392), Funds of Science Technology Department
of Zhejiang Province (grant nos. 2012C13021-2 and 2012C37103),
Zhejiang Leading Team of S&T Innovation (grant no. 2011R50015)
and the Fund of Health Bureau of Zhejiang Province (grant no.
2010SSA006).
References
|
1
|
Yang C, Yang J, Sun M, Yan J, Meng X and
Ma T: Alantolactone inhibits growth of K562/adriamycin cells by
downregulating Bcr/Abl and P-glycoprotein expression. IUBMB Life.
65:435–444. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Melo JV and Chuah C: Novel agents in CML
therapy: Tyrosine kinase inhibitors and beyond. Hematology Am Soc
Hematol Educ Program. 427–435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carafa V, Nebbioso A and Altucci L:
Histone deacetylase inhibitors: Recent insights from basic to
clinical knowledge & patenting of anti-cancer actions. Recent
Pat Anticancer Drug Discov. 6:131–145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lemal R, Ravinet A, Moluçon-Chabrot C, Bay
JO and Guièze R: Histone deacetylase inhibitors in the treatment of
hematological malignancies. Bull Cancer. 98:867–878. 2011.(In
French). PubMed/NCBI
|
|
5
|
O'Connor OA, Heaney ML, Schwartz L,
Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C,
Portlock C, Horwitz S, et al: Clinical experience with intravenous
and oral formulations of the novel histone deacetylase inhibitor
suberoylanilide hydroxamic acid in patients with advanced
hematologic malignancies. J Clin Oncol. 24:166–173. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kelly WK and Marks PA: Drug insight:
Histone deacetylase inhibitors-development of the new targeted
anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract
Oncol. 2:150–157. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yamamoto S, Tanaka K, Sakimura R, Okada T,
Nakamura T, Li Y, Takasaki M, Nakabeppu Y and Iwamoto Y:
Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or
autophagy-associated cell death in chondrosarcoma cell lines.
Anticancer Res. 28:1585–1591. 2008.PubMed/NCBI
|
|
8
|
Nimmanapalli R, Fuino L, Stobaugh C,
Richon V and Bhalla K: Cotreatment with the histone deacetylase
inhibitor suberoylanilide hydroxamic acid (SAHA) enhances
imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia
cells. Blood. 101:3236–3239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fiskus W, Wang Y, Joshi R, Rao R, Yang Y,
Chen J, Kolhe R, Balusu R, Eaton K, Lee P, et al: Cotreatment with
vorinostat enhances activity of MK-0457 (VX-680) against acute and
chronic myelogenous leukemia cells. Clin Cancer Res. 14:6106–6115.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu C, Rahmani M, Conrad D, Subler M, Dent
P and Grant S: The proteasome inhibitor bortezomib interacts
synergistically with histone deacetylase inhibitors to induce
apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571.
Blood. 102:3765–3774. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo N and Peng Z: MG132, A proteasome
inhibitor, Induces apoptosis in tumor cells. Asia Pac J Clin Oncol.
9:6–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jagani Z, Song K, Kutok JL, Dewar MR,
Melet A, Santos T, Grassian A, Ghaffari S, Wu C, Yeckes-Rodin H, et
al: Proteasome inhibition causes regression of leukemia and
abrogates BCR-ABL-induced evasion of apoptosis in part through
regulation of forkhead tumor suppressors. Cancer Res. 69:6546–6555.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller CP, Ban K, Dujka ME, McConkey DJ,
Munsell M, Palladino M and Chandra J: NPI-0052, a novel proteasome
inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and
in combination with HDAC inhibitors in leukemia cells. Blood.
110:267–277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tong Y, Liu YY, You LS and Qian WB:
Perifosine induces protective autophagy and upregulation of ATG5 in
human chronic myelogenous leukemia cells in vitro. Acta
Pharmacol Sin. 33:542–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Park WH and Kim SH: MG132, a proteasome
inhibitor, induces human pulmonary fibroblast cell death via
increasing ROS levels and GSH depletion. Oncol Rep. 27:1284–1291.
2012.PubMed/NCBI
|
|
17
|
Heaney NB and Holyoake TL: Therapeutic
targets in chronic myeloid leukaemia. Hematol Oncol. 25:66–75.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marks PA, Richon VM and Rifkind RA:
Histone deacetylase inhibitors: Inducers of differentiation or
apoptosis of transformed cells. J Natl Cancer Inst. 92:1210–1216.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gorre ME and Sawyers CL: Molecular
mechanisms of resistance to STI571 in chronic myeloid leukemia.
Curr Opin Hematol. 9:303–307. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shah NP, Nicoll JM, Nagar B, Gorre ME,
Paquette RL, Kuriyan J and Sawyers CL: Multiple BCR-ABL kinase
domain mutations confer polyclonal resistance to the tyrosine
kinase inhibitor imatinib (STI571) in chronic phase and blast
crisis chronic myeloid leukemia. Cancer Cell. 2:117–125. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sankari SL, Masthan KM, Babu NA,
Bhattacharjee T and Elumalai M: Apoptosis in cancer-an update.
Asian Pac J Cancer Prev. 13:4873–4878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Azad MB, Chen Y and Gibson SB: Regulation
of autophagy by reactive oxygen species (ROS): Implications for
cancer progression and treatment. Antioxid Redox Signal.
11:777–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ling LU, Tan KB, Lin H and Chiu GN: The
role of reactive oxygen species and autophagy in safingol-induced
cell death. Cell Death Dis. 2:e1292011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stein SJ and Baldwin AS: NF-κB suppresses
ROS levels in BCR-ABL(+) cells to prevent activation of JNK and
cell death. Oncogene. 30:4557–4566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nguyen T, Dai Y, Attkisson E, Kramer L,
Jordan N, Nguyen N, Kolluri N, Muschen M and Grant S: HDAC
inhibitors potentiate the activity of the BCR/ABL kinase inhibitor
KW.2449 in imatinib. sensitive or. resistant BCR/ABL+leukemia cells
in vitro and in vivo. Clin Cancer Res. 17:3219–3232. 2011.
View Article : Google Scholar : PubMed/NCBI
|